
Volume 22 Number 2
How to integrate new technologies into daily practice: The clinician perspective
Luc Téot, Sylvie Meaume, Jean Charles Kerihuel
Keywords wound healing, regulations, Keywords: Technology, clinical trials
DOI 10.35279/jowm202107.02
Abstract
The integration of new technologies can call for obeying different rules and should be considered a long-term evolutionary process that is realised step by step. Before a medical device appears in the market, a long regulatory process has been completed, including safety, experimental and preclinical studies leading to an authorisation for distribution in a national market. Depending on the target countries and their respective regulations, the need for robust clinical studies, including randomised clinical trials, is often mandatory for distribution. Post-clinical trials may be used to confirm the results obtained from a medical device. The efficacy of the introduction of new technology in daily practice is then dependent on clinicians’ perceptions of the improvement in wound-healing. Complications may occur even after a long period of time, with severe consequences for patients’ quality of life. Since the PIP (Poly Implant Prothèse) scandal, more restrictions have been imposed to prevent such failures and increase the safety for the population.
INTRODUCTION
Changing practices is a difficult process in wound healing, as new technologies regularly emerge, forcing experts to create a new space for integrating these devices into their practical armamentarium.
This process has accelerated dramatically in the past 50 years. Indeed, we are currently facing new conceptual revolutions with the emergence in daily practice of machine learning devices and the development of artificial intelligence-based medicine, in addition to new therapeutic applications of gene-editing technologies. These technological introductions require changes in our knowledge acquisition processes and in patient–health professional relationships, to successfully address potentially dramatic challenges.
Introducing any new technology into daily practice requires careful thinking and precise planning. Several steps must be followed; this process may help to inform learning by using the most efficient means available.
Prerequisites before introducing a new technology
The authorisations to use medical devices given by national authorities are not subject to the same level of scrutiny that drugs are, in terms of critical levels of evidence-based medicine (EBM), but many regulatory departments and/or reimbursement agencies believe that companies proposing new medical devices (MD) should bring a high level of EBM when asking for reimbursement.
One of the first questions to be asked in this process concerns the class of the new medical device, as defined by the European Community Medicine Evaluation committee (MEDEV).
With the above in mind, designing the right protocol has become an art that calls for following specific guidance, as comparing two groups presenting identical wound-healing pathologies and outcomes remains extremely difficult. Developing randomised controlled trials (RCTs) is cost-prohibitive for most companies, and the risks associated with failing to show statistical differences between the study groups remains high. This is why so many new technologies are sustained by secondary evidence. Most of the time, a consensus can be obtained among key opinion leaders, which is sufficient enough, in some countries, to be used as a convincing argument.
At this point, fundraising to develop a solid RCT can be planned. Even with promising results and publication in a high impact factor journal, obtaining a ‘green light’ from the national authority to penetrate the market remains to be done. Companies should strive to add to physicians’ and practitioners’ knowledge using a strategy of diffusing good information and beginning the real work of influencing prescribers. Post-marketing studies are often developed to complete the evidence and add more practical information, or to extend the device’s clinical indications, which are often very focused during the first RCT.
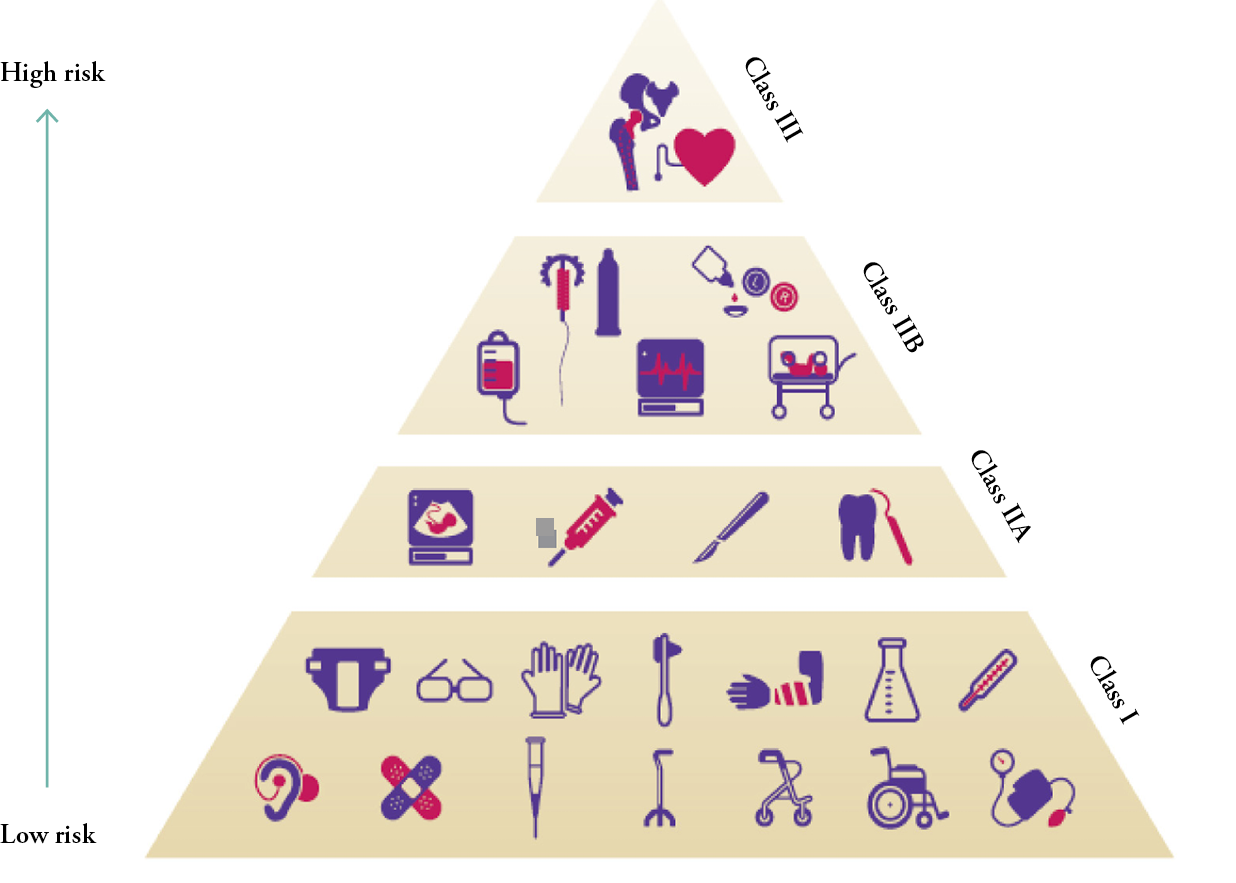
Figure 1: Most of the dressings that contain no active products are in Class I.
Those with less than 10% active products are classified as Class IIA or IIB.
Regulations
a) Generalities
When assessing the state of medical device regulations in Europe and the United States (US), the US regulations seem to be better arranged, which is likely due to the fact that there is only one responsible body — the US Food and Drug Administration, which is responsible for all medical device regulations. By contrast, in the European Union (EU), new regulations are proposed by commissions composed of members coming from the 27 EU member states, and it may take a long time before all EU countries come to an agreement.1 Another difference in the decision-making process concerns the levels and chains of decision making, from physicians proposing new devices to health policymakers and the weight of regulations. In other words, an open market leaves more opportunities to develop and adopt new technologies than a centralised one does, as it will be less open to the introduction of new sources of expenses to be paid by the public insurance system. Regulations remain a political issue in many countries around the world. In Europe, even if a European Certification approval is obtained, clinical trials must be developed based on a methodology that is more or less rigorous, depending on the requirements of each national agency.
b) The device approval process
In Europe, most of the health agencies do not allow any medical devices to come to market before receiving CE approval, a proof of biocompatibility, completed by the national health agency’s authorisation process. Medical devices make an essential contribution to healthcare in the EU for the benefit of European citizens. From sticking plasters to X-ray scanners, dentures, hip joints and in-vitro diagnostic devices that monitor diabetes or identify infections, medical devices are crucial for diagnosing, preventing, monitoring and treating illness, and for overcoming disabilities. They are also important to the economy, providing €110 billion in sales and 675,000 jobs in Europe in 2020. The EU is a net exporter in this sector.
European legislation ensures the safety and efficacy of medical devices and facilitates patients’ access to devices in the European market. To keep up with advances in science and technology, two new European regulations are replacing three existing directives in the years leading up to 2022.
c) Regulatory frameworks
Regulations have established a modern, robust EU legislative framework for ensuring the protection of public health and patient safety, which has boosted consumer confidence in the medical devices industry. Medical devices in the EU have been driven since 1993 by Council Directive 93/42/EEC, and on 5 April 2017, two new regulations on medical devices and in vitro diagnostic medical devices were adopted. The new regulations for medical devices, Regulation (EU) 2017/745 of the European Parliament and the Council of 5 April 2017 on Medical Devices, which amended Directive 2001/83/EC, Regulation (EC) No. 178/2002, went into full effect in May 2020. The new regulations contain a series of improvements to modernise the current system (Table 1).

The main reasons behind the new regulations for medical devices were linked to problems with diverging interpretations of the previous directives and an incident concerning the fraudulent production of PIP (Poly Implant Prothèse) silicone breast implants in 2010, which highlighted weaknesses in the legal system in place at the time and damaged the confidence of patients, consumers and healthcare professionals concerning the safety of medical devices. Such problems should not occur again, and the safety of all medical devices available in the EU had to be strengthened. Moreover, a revision of the legislation was necessary to consolidate the role of the EU as a global leader in the sector over the long term and to take into account all technological and scientific developments in the sector.
What are the consequences of these new regulations for wound care practices?
Will these improvements, by imposing more administrative constraints, accelerate or decrease the translation of research developments? Will the whole process result in an earlier selection of the best technologies, thus guaranteeing a higher gain for the patients? Will the companies need to change their modus operandi? Medical devices in wound care are still suffering from a poor image compared to those developed for treating cardiac diseases, rheumatology and orthopaedics. Reimbursement prices are also comparatively lower for wound care products, a circumstance that is probably linked to the poor global knowledge of not only wound healing among decision-makers, but also of market needs and the difficulties of developing adapted RCTs. In this direction, a deeper influence of experts in the field is needed.
From approval to market diffusion
Once the CE mark has been obtained, companies must follow three successive steps:
a) Approval by national authorities. Safety rules are specific to each country related to any new chemical product acting either as an absorber, a debrider or having the capacity to enhance epidermisation, and there are specific national regulations concerning the percentage of active drugs or compounds that may be included in a device. It is beyond the scope of this paper to detail the international regulations of the European Community and the US concerning the development of new medical devices, their assessment, pricing and reimbursement of medicines, which were proposed by the MEDEV (medev-com.eu) since 1998.
At this time, during the step 1 of this process, companies may choose to try to penetrate a market without reimbursement, based on a strategy of marketing and promoting the device. Key opinion leaders give expert opinions, reach consensus and hold conferences, and the support of users is gained through a direct approach, confirmed by representatives of the MD companies. Medico-economic studies may provide complementary arguments capable of convincing the payers to support new devices.
b) In some countries, a double regulatory framework and an evaluation of added value is needed. In some countries, a commission charged with evaluating the added value of new devices is contacted when reimbursement is in focus. This commission will assess the evidence of any superiority to the presently used MD, if an RCT confirms this point. In some countries, the need for a statistical superiority may be replaced by a temporary authorisation for reimbursement by submitting to an evaluation of the MD’s use during a determined period of time. This commission may determine if the device is new and should be considered an MD registered under a brand name, or if the MD is comparable to already existing devices and should be considered a generic product.
c) Pricing. A second commission must determine the public reimbursement price, if any, by analysing the company’s proposals and dealing with them to set a price that will be adopted for the country. In France, until 2019, this price was only considered ‘advice’ to pharmacists, who were free to follow it or to determine their own pricing, but this has recently been transformed into a mandatory directive so that the final public price cannot be modified. This strict price-setting is more pronounced in countries where the reimbursement rate is highly regulated because of the social insurance system. In other countries, price-setting is not applied and prices are set freely.
Which questions should physicians ask before adopting a new technology?
In healthcare systems, both in hospitals and private practices, three domains can be schematically involved by innovations: therapeutic, diagnostic and managerial/organisational. In each situation, two types of innovation may be considered: conceptual and contextual.
The first type concerns the introduction of a very new technology at the preliminary stage of its lifecycle. Practical experiences with this technology are still limited, and a wide range of questions should be raised.
By contrast, contextual innovations concern technologies that are more or less at a mature stage of their lifecycle. In this case, a great deal of benefit–risk and cost–utility evidence is often available, and the processes followed to decide on a device’s introduction and to plan its implementation are simpler.
Nevertheless, regardless of the nature of the innovation (conceptual or contextual), a similar checklist of topics should be considered (Table 2).

The pressure to innovate
None of the questions detailed above are easy to answer. In addition, answers might vary according to cultural contexts and the economic circumstances of the concerned country.
One of the most dangerous problems with new technologies is that they are new. Just as consumers are confronted by high-tech innovations via social network channels, healthcare professionals are under pressure from many actors, including manufacturers, opinion leaders and even patients themselves. It is difficult to resist the novelty of new technologies, and often, like a mutated virus crossing species barriers, they soon spread worldwide and invade communities that are not ready to implement their resulting changes appropriately.
Indeed, answering the above questions is a time-consuming process. It takes months, and often years, to build evidence-based health technology assessment reports capable of guiding professionals in their decisions, and years are the most terrific enemy of novelty.
Another valid question is how we can integrate artificial intelligence into scalable production plans. One new field of interest in medicine has been the introduction of deep learning machines into the definition of clinical situations based on images. In radiology, to detect early tumours, and in dermatology, to diagnose melanomas, some software can now offer help with the decision-making process, based on millions of images that form a database and feed a neural network software. The use of such technology in wound healing will not only necessitate the building of similar databases, but also the asking of the right questions based on strong collaboration between clinicians and engineers/mathematicians. This solution is technical.
When it comes to genetic therapies, other constraints must be respected, such as the risk of malignancy after genetic manipulation. Bioethics committees in each country are working closely with concerned clinicians to discuss and establish regulations. This solution is political.
Examples of recommendations on methodologically key issues for adopting a new technology
1) Some authors5 have recently identified success factors for the effective implementation of new technologies and technological equipment in operating rooms (ORs), based on a systematic literature review. They analysed 10 databases and reviewed their included articles. The search resulted in 1592 titles for review, and 37 articles for final inclusion in the study. Influencing factors were separated from resulting factors based on the outcomes. Six main categories of influencing factors on the successful implementation of medical equipment in ORs were identified:
- Processes and activities
- Staff
- Communication
- Project management
- Technology
- Training
A seventh category, performance, referred to resulting factors during implementations.
Greenhalgh et al.3 considered that aligning the identified influencing factors during implementation impacts the success, adaptation and safe use of new technological equipment in the OR, and thus the outcome of an implementation.
2) More generally, the lifecycle of a new technology, reasons for adoption, non-adoption and abandonment have been described4 and can be summarised as indicated below (see Figure 2).
Figure 2: Steps for adopting a new technology, from Mytton et al.5
Complications stemming from the poorly managed introductions of new medical devices without enough evidence
a) The example of metal-on-metal (MoM) hip replacements
New medical technologies are often used widely without adequate supporting data, a practice that can lead to widespread catastrophic failure, as occurred with metal-on-metal (MoM) hip replacements. Hunt et al.6 determined both how revision rates would have differed if, instead of receiving MoM hip replacements, patients had received existing alternatives, and the subsequent cumulative re-revision rates of patients who received MoM hip replacements compared with alternatives.
Their study focused on a population-based longitudinal cohort of patient data recorded in the National Joint Registry (NJR) for England, Wales and Northern Ireland between April 2003 and December 2014. The authors ascertained implant failure rates separately among stemmed MoM total hip replacement (THR) and hip-resurfacing procedures and, using flexible parametric survival modelling, compared them with the failure rates that would have been expected had existing alternatives been used. They used a Kaplan-Meier survivorship analysis to compare the cumulative re-revision rates of patients who received stemmed MoM primary replacements that failed to those who underwent hip resurfacing that failed after using non-MoM THRs.
In all, 37,555 patients underwent MoM hip resurfacing, with a 10-year revision rate of 12.6% (95% confidence interval [CI]: 12.2–13.1%), compared with a predicted revision rate of 4.8% if alternative implants had been used. The 32,024 stemmed MoM THRs had a 19.8% (95% CI: 18.9–20.8%) 10-year failure rate, compared with an expected rate of 3.9% if alternatives had been used. For every 100 MoM hip-resurfacing procedures, there were 7.8 excess revisions by 10 years, and for every 100 stemmed MoM THR procedures, there were 15.9, which equates to 8,021 excess first revisions. Seven-year re-revision rates were 14.9% (95% CI: 13.8–16.2%) for stemmed non-MoM THRs, 18.0% (95% CI: 15.7–20.7%) for MoM hip resurfacing and 19.8% (95% CI: 17.0–23.0%) for stemmed MoM THRs.
This study highlights the consequences of a widespread and poorly monitored adoption of a medical technology. More than 1 million MoM hip prostheses were implanted worldwide, so the excess failure on a global scale was enormous. This practice of adopting new technologies without adequate supporting data must not be repeated.
b) In the wound care field, the implementation of NPWT (negative pressure wound therapy) is a clear example of this problem, but with less dramatic consequences. This technology has been largely implemented without any strong evidence of its efficacy demonstrated in RCTs. The principle of NPWTs’ action is double; one is linked to a specific foam (pore size and capacities of extracting liquids under pressure) and the other is aspiration. Most users, mainly surgeons, were convinced of the approach and used it in many clinical indications, applying the principle that ‘when it works in practice, the theory will follow’. However, the lack of high-level RCTs demonstrating the added value of the foam had two long-term consequences, the difficulty of obtaining an adapted reimbursement and the introduction by concurrent companies of devices focused solely on aspiration, which led to different clinical results and controversies in the user community. Surgeons claimed the foam’s spectacular efficacy and the simplification in wound care management, and the tool was adopted without being subject to a methodical decision process. Evidence-based assessments came later, but were considered by some statisticians to be biased, and consensus was hard to reach. While still a topic of debate, accumulated pragmatic field evidence suggests that the spread of NPWT was not a mistake.2 Nevertheless, this type of chaotic introduction of new technologies in daily practice should be avoided, as the challenges in the ensuing years are so dramatic in many aspects that there is no room for empiricism.
The compliance of physicians in adopting a new technology
In a recent study by Ruiz Morilla et al.7 to evaluate the opinion of physicians regarding e-health, a questionnaire they had previously designed and validated was used to interview 930 physicians. The usefulness of telemedicine scored 7.4 (SD 1.8) on a scale from 1–10 (lowest to highest), and the importance of the Internet in the workplace was rated at 8.2 (SD 1.8). Therapeutic compliance (7.0, SD 1.8) and patient health (7.0, SD 1.7) showed the best scores, but there were differences between professionals who had or had not previously participated in a telemedicine project (p < 0.05). Physicians believe in the usefulness of e-health. Professionals with previous experience with it are more open to its implementation and consider that the benefits of technology outweigh its possible difficulties and shortcomings. The relationship of users to technology differs according to their personal or professional experiences.
In contrast to the above, some MDs that have obtained a certain level of evidence and whose efficacy has been commented on in journals with high impact factors, like honey or maggots, still suffer from mental projections in users’ minds. Honey is not generally regarded as innovative, and maggots are often linked with images of post-mortem decomposition, an image stronger in Southern Europe. These examples highlight the other factors that may impact a device’s image of innovation and marketers’ and lobbyists’ ability to renew the use of an old medical device.
CONCLUSION
Implementing new technologies in daily practice is never an easy task, despite it being necessary for the promotion of efficient healthcare management. The help of various professionals not always based in the medical field is also sometimes needed. Each individual must bring their own contribution, but a new medical device has a greater chance of being successful if professionals are involved in its development from the beginning. Technical achievement is also crucial for remaining in compliance with all regulatory requirements. One of the most difficult steps is the building, developing and analysing of the results of a properly realised RCT. Another is ensuring a device’s adoption by a wide number of professionals.
Author(s)
Luc Téot
MD, PhD, Montpellier University Hospital, Cicat Occitanie Hôpital La Colombière CHU de Montpellier 38 rue Charles Flahaut
34295 Montpellier cedex 5
Sylvie Meaume
MD, APHP Paris, Rothschild Hospital
Jean Charles Kerihuel
MD. Vertical Paris
Correspondence: l-teot@chu-montpellier.fr
Conflicts of Interest: None
References
- Kacetl J, Maresova P. Legislative and ethical aspects of introducing new technologies in medical care for senior citizens in developed countries. Clin Interv Aging. 2016; 11: 977.
- Téot L, Guillot-Masanovic M, Miquel P, Truchetet F, Meaume S, Dompmartin A, et al. Clinical impact of negative-pressure wound therapy: A 1126-patient observational. Wound Repair Regen. 2014; 22(3):341–50. doi: 10.1111/wrr.12179.
- Greenhalgh T, et al., Beyond adoption: A new framework for theorizing and evaluating nonadoption, abandonment, and challenges to the scale-up, spread, and sustainability of health and care technologies. J Med Internet Res. 2017; 19(11):e367.
- Mytton OT, Velazquez A, Banken R, Mathew JL. Ikonen TS, Taylor K, et al. Introducing new technology safely. Qual Saf Health Care. 2010; 19(Suppl 2):i9ei14. Doi:10.1136/qshc.2009.038554.
- Misser NS, van Zaane B, Jaspers JEN, Gooszen H, Versendaal J. Implementing medical technological equipment in the OR: Factors for successful implementations. J Healthc Eng. 2018. doi.org/10.1155/2018/8502187.
- Hunt LP, Whitehouse MR, Beswick A, Porter ML, Howard P, Blom, AW. Implications of introducing new technology: Comparative survivorship modeling of metal-on-metal hip replacements and contemporary alternatives in the National Joint Registry. J Bone Joint Surg (Am ed); 100(3):189–96. https://doi.org/10.2106/JBJS.17.00039.
- Ruiz Morilla MD, Sans M, Casasa A, Giménez N. Implementing technology in healthcare: Insights from physicians. BMC Medical Inform Decis Mak. 2017; 17:92. doi 10.1186/s12911-017-0489-2.