

Volume 25 Number 4
Altered macrophage phenotypes impair wound healing
Katherine B Sahin, Mark Hesketh, Zoe E West and Rachael Z Murray
Keywords Inflammation, macrophage, M1 and M2, wound.
Abstract
One of the defining features of chronic wounds is their high levels of inflammation. Patients present with high levels of inflammation, around 80%, of cells at the wound margin being macrophages and with wound fluid laden with pro-inflammatory cytokines. The latter is in part responsible for preventing wound closure, along with the low levels of growth factors. In healthy acute wounds, two types of macrophages can be found: pro-inflammatory M1 macrophages and anti-inflammatory M2 macrophages, with more M1 cells present early post-injury and M2 appearing later to regulate repair and wound closure. This pro-repair M2 phenotype, that secrete a range of mediators including growth factors that regulate re-vascularisation and closure of a wound, is lacking in chronic wounds, leading to excessive inflammation, enhanced degradation within the wound, reduced matrix deposition and lack of closure. This review will discuss the alterations seen in chronic wound macrophages, how this might affect the repair process and some potential reasons for their dysregulation.
Introduction
Most wounds heal or at least close relatively quickly, but for some people wounds can take much longer to close, with wounds that do not heal in a timely manner (4–6 weeks) being defined as chronic1. These wounds typically occur on the lower limbs of mainly ageing patients, many of whom have an underlying chronic disease, such as diabetes or chronic venous insufficiency, that can contribute to wound chronicity. Impaired healing can last from months to years and have an enormous impact on patient quality of life2. In addition to being painful, wounds are often malodorous, can result in loss of mobility and social isolation, leading to depression in some cases3. Treatments can be unreliable with their outcomes often unpredictable. Worldwide, complications from these chronic wounds lead to one major amputation every 30 seconds, which amounts to over 2500 limbs lost a day, with one in four amputees requiring contralateral amputation and/or re-amputation. For diabetic foot ulcer patient amputees the mortality rate is high, at 39–80% five years post-amputation4. Compare this to the 10% mortality rate for breast cancer patients at five years (data from 2009–2013) post-diagnosis then this figure is alarming. The financial impact of chronic wounds is also substantial, with wound management accounting for around 3% of the total health care expenditure in developed countries. It is estimated that $2.85 billion per annum is spent on chronic wounds in Australia alone and with the increasing age and population with diabetes, the number of patients with chronic wounds is expected to rise dramatically5. While in-roads have been made into understanding the repair process, there is still much to understand about why these wounds occur, how they differ from an acute wound and how they might be altered to improve wound healing. Improved understanding will aid in the targeting of therapies to prevent or speed up healing of chronic wounds.
Repairing skin
Wound healing is a highly complex, multistage process that starts the instant an injury occurs. It involves skin cells such as keratinocytes, fibroblasts, endothelial cells, the recruitment of immune cells, the laying down of new extracellular matrix (ECM) components that act as a scaffold for these cells, and a variety of soluble mediators that coordinate the process6. It can be divided into four overlapping phases: haemostasis, inflammation, proliferation, and then maturation. Upon injury, blood vessels constrict to reduce blood flow and to seal the damaged vessel. Platelets adhere together in a fibrin matrix to form a clot that provides a scaffold that will support structures necessary for vessel repair and migration of immune cells into the wound. Studies in mice show that within minutes microhaemorrhaging into the wound allows a small pool of neutrophils and macrophages to access the wound7,8. Soluble factors released from platelets then recruit more neutrophils to the wound, with numbers peaking around days 1–2 post-injury8. These cells are the first line of defence and their role is to phagocytose (eat and destroy) pathogens, dead cells and debris, thus cleaning the wound, and to phagocytose dead cells and debris to debride the wound, and secrete soluble mediators that will attract other immune cells to the wound. Neutrophils have a very short life span and, within a day or two, undergo a form of programmed cell death known as apoptosis at the site of injury. These spent neutrophils need to be removed and macrophages fulfil this role by phagocytosing the neutrophils. Importantly, the phagocytosis of neutrophils alters the function of the macrophage, pushing them towards being more reparative than inflammatory in their phenotype. This phagocytic function and switching to the reparative phenotype has been shown in diabetic wounds to be inhibited, resulting in wounds that remain in the inflammatory stage that will not heal. This impairment in function is seen in both the diabetic mice models and in diabetic wounds of patients. This will be discussed below.
The majority of macrophages found in wounds are recruited from the bone marrow and blood into tissue as monocytes, their precursors. As these monocytes exit the blood, they migrate into injured tissue. Factors in the local environment then cause them to differentiate into macrophages8. Their numbers in an acute wound increase until day 2 and then stay at a steady level until 5 days post-injury, when numbers progressively decrease, so that by day 14 few macrophages remain8. In addition to removing spent neutrophils, the macrophage has numerous roles in the wound as they secrete a plethora of cytokines, chemokines, growth factors and other mediators. In the early stages, they secrete factors that increase inflammation and recruit more macrophages; later they secrete mediators that regulate the proliferative stage. Growth factors and other mediators released by macrophages trigger fibroblasts, keratinocytes and endothelial cells to proliferate, migrate, and differentiate. Fibroblasts differentiate into myofibroblasts that lay down new extra cellular matrix (ECM), such as fibronectin and collagen III, that form the framework for cells to enter and populate the wound. Endothelial cells create new blood vessels and a thin layer of keratinocytes covers the open wound, in a process known as re-epithelialisation. The remodelling and maturation stage occurs once the wound is closed. The ECM is continuously degraded and restructured to increase the strength of the new tissue and the wound contracts to reduce size9. During this time, macrophages produce enzymes that aid this remodelling of the ECM. Collagen I is substituted for collagen III, resulting in the formation of the final scar10. This final phase can take years to fully complete, depending on the size, severity and healing time of the wound. The scar formed here, in the case of healed chronic wounds, can be quite fragile.
Changes in macrophage phenotypes alter the course of healing
As mentioned above, wound macrophages play a range of roles and regulate the function of numerous other cells in the wound. These functions need to be coordinated in a timely and sometimes sequential fashion. Macrophages do this by altering their phenotype through cues in the wound environment. Broadly speaking, macrophages have been classified as pro-inflammatory M1 or anti-inflammatory/reparative M2 macrophages (Table 1)11-13. This classification is an oversimplified one as multiple M2 phenotypes exists, although not all exist in wounds. In vivo, there is a continuum of phenotypes between M1 and M2 that evolve as wound healing progresses (Figure 1). M1 macrophages, found in the early stages post-wounding, are activated by stimulants in the local wound environment; this can include interferon-g (IFN-g), tumour necrosis factor (TNF) and damage-associated pattern molecules (DAMPs). The latter are biomolecules released by damaged ECM and injured cells that act as endogenous danger signals to tell the immune system that an injury has occurred. These M1 macrophages are prolific producers of pro-inflammatory cytokines, such as TNF and interleukin-6 (IL-6), and other mediators that regulate the early stages of wound healing (Table 1). They are also proficient phagocytes and engulf spent neutrophils. This phagocytosis step, known as efferocytosis, along with environmental factors (IL-4 or IL-13) can push M1 macrophages into an M2 anti-inflammatory/reparative phenotype. M2 macrophages can be separated in vitro into four phenotypes, M2a-d, based on their stimulation, surface markers and the cytokines, chemokines, growth factors and other mediators they produce and, in most cases, secrete (Table 1). M2 macrophages produce anti-inflammatory cytokines that act to switch off inflammation in the wound. They also act as a master regulator of repair by producing various growth factors, such as vascular endothelial growth factor (VEGF) that regulates formation of new blood vessels, and transforming growth factor-b (TGF-b), which plays a key role in regulating proliferation, migration, differentiation, and ECM production during the proliferation and maturation phases of repair.
Table 1: Macrophage phenotypes.
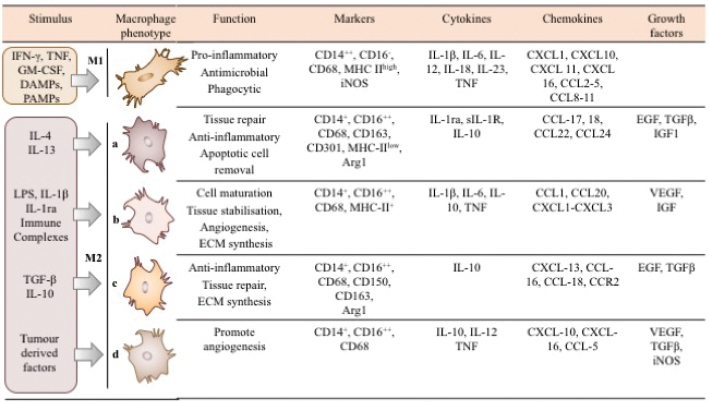
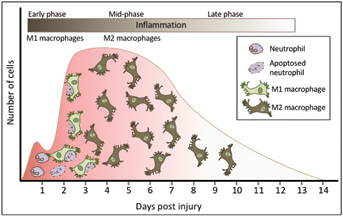
Figure 1: Macrophage phenotypes alter over the course of wound healing. In the early stages, an acute wound is populated with approximately 80% M1 pro-inflammatory macrophages but as the wound mature these macrophages swap phenotype to become M2 anti-inflammatory macrophages that are pro-repair.
Macrophages are dysregulated in chronic wounds
In an acute wound at the start of the inflammatory phase, approximately 85% of macrophages have an M1 pro-inflammatory phenotype but by five days post-injury only 15% of the macrophages found in the wound are M1 macrophages14,15. In chronic wounds this shift in phenotypes does not occur and approximately 80% of the cells found at the chronic wound margin are macrophages with an M1 pro-inflammatory phenotype16-19 (Figure 2). As a result, patients present with high levels of pro-inflammatory cytokines, such as TNF and IL-6, as well as iNOS in their wound fluid17,20,21. These high levels of TNF and other pro-inflammatory mediators leads to the recruitment of more immune cells and it disrupts both the levels and delicate balance between matrix metalloproteinases (MMPs) and their inhibitors, the tissue inhibitor of metalloproteinases (TIMPs)22. In an acute wound these MMPs play a role in the remodelling of the ECM and their activity is finely tuned by their inhibitors, the TIMPs. However, when these MMPs have free reign, as seen in chronic wounds, either by their overexpression and/or a reduction in their inhibitors, their excess proteolytic activity leads to the destruction of the ECM scaffold that cells require to repopulate and close the wound22. This impaired wound healing has been studied in mice and the addition of recombinant TNF into mouse wounds delays the repair process and leads to senescence of fibroblasts17. A number of therapeutic antibodies that inhibit TNF exist including Infliximab and Adalimumab. When used topically (Infliximab) or subcutaneously (Adalimumab) in chronic venous ulcers (CVU) of patients whose wounds failed to respond to any previous conventional treatment it improved wound closure23,24. Although these studies were limited in that they had no placebo-treated patients, their results mimicked results seen in mouse studies where inhibiting TNF improved impaired wound healing in diabetic mice compared to control mice17.
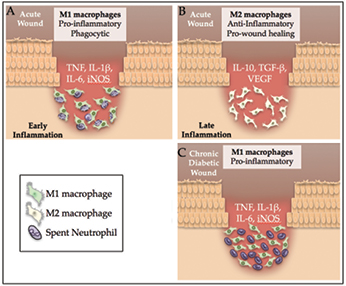
Figure 2: Chronic wounds from patients with diabetes have high levels of inflammation and few M2 macrophages compared to an acute wound. A) In the early stages of repair of an acute wound, factors in the wound environment activate macrophages to differentiate into pro-inflammatory M1 macrophages. These M1 macrophages are prolific secretors of pro-inflammatory cytokines and they diligently phagocyte apoptosed neutrophils. B) The phagocytosis of spent neutrophils pushes M1 macrophages to differentiate and form pro-repair, anti-inflammatory M2 macrophages. C) In a diabetic chronic wound, macrophages are unable to phagocytose the spent neutrophils and so remain in a chronic inflammatory state that prevents wound closure with few M2 macrophages present.
The detrimental effects of TNF are compounded by the lack of M2 macrophages. These cells are key drivers of the proliferative stage, and so chronic wound fluid contains much lower levels of growth factors, such as TGFb1, VEGF and insulin-like growth factor-1 (IGF-1), than would be expected19,25. In an acute wound, the number of cells with an M2 phenotype normally increases as the wound matures. Using M2 macrophages transplanted from the later (day 5) proliferative stages to the earlier inflammatory stage (day 3) it has been shown that these M2 cells increase fibroblast proliferation and vascular regeneration15.
One of the big questions is then why are chronic wounds populated by an excess of M1 macrophages with very few M2 macrophages in the wound? Studies using the cFMS kinase inhibitor GW2580 to block the M1 to M2 switching of phenotypes in acute wounds show that failure to switch to M2 lengthens the inflammatory stage, suggesting defects in the switch might contribute to the protracted inflammation seen in chronic wounds26. In vitro M2 macrophages can be formed by stimulating macrophages with certain cytokines, such as IL-4, IL-10 or IL-13 (Table 1). However, in an acute wound where macrophages switch from M1 to M2 quite readily, little extracellular IL-4 and IL-13 has been found14. In fact, the addition of M2a or M2c macrophages, formed in vitro by stimulation with IL-4 or IL-10 respectively, to wounds had little effect on wound closure in mice, suggesting that IL-4, IL-10 and IL-13 may not play a role in driving this switch27. One other way to drive the M1 to M2 switch in macrophages is by their phagocytosing dead neutrophils that have apoptosed28-30. In vitro, cells formed this way are of an M2b macrophage phenotype that produces high levels of IL-10 and TGF-b31. Wounds are typically populated with spent neutrophils in the early stages of repair and their clearance by macrophages could drive the switch from M1 to M232. Therefore, the macrophage phagocytic ability, or lack of, could ultimately alter whether a wound will remain in a pro-inflammatory state or heal in a timely manner.
Unlike macrophages from acute wounds, cells taken from patients with diabetes with chronic wounds show these macrophages have a defect in their ability to phagocytose apoptosed neutrophils33 (Figure 2). This deficit is a result of hyperglycaemia and its associated advanced glycated end products (AGEs), which alter the macrophages ability to phagocytose prior to them entering the wound34,35. These macrophages are also activated prior to entering the wound rather than by factors in the wound. This inability to phagocytose increases the number of apoptotic cells in these wounds and inhibits the M1 to M2 switch in phenotype, generating more inflammation36. What then might be responsible for the defective M1 to M2 switch in other types of chronic wounds? Chronic venous disease patients present with venous hypertension in lower limb deep and superficial veins, which results in changes to the endothelium and macrophage activation. Consequently, as seen with patients with diabetes, macrophages in these patients are altered prior to entering the wound and there is low-level chronic inflammation prior to the wound forming. Alterations are due to the chronic inflammation and to their phagocytosing red blood cells that have leaked into tissue and increasing their intracellular iron levels. Iron-loaded macrophages taken from the wound margin of iron-dextran-treated mouse models at five days post-injury show that these cells are predominantly of a pro-inflammatory M1 phenotype with an intermediate anti-inflammatory M2 phenotype (TNF-αhi, IL-12hi, CCR2hi, Ly6Chi, Dectin-1med, IL-4Rαmed, and CD204med), suggesting that they do not form the typical M2 macrophages17. In iron-dextran-treated mice removal of iron with the chelator desferrioxamine improved wound healing, suggesting elimination of iron might improve these wounds17. Whether these iron-laden macrophages have a defect in phagocytosis is yet to be determined, but patients present with approximately 80% of wound margin cells being predominantly M1 macrophages and few M2 macrophages similar to that seen in diabetic ulcers17. Like diabetic wounds, the increased and prolonged M1 macrophage phenotype results in a pro-inflammatory state that is detrimental to wound healing.
Summary
Chronic wounds, regardless of the aetiology, are characterised by an increased and protracted inflammatory stage, with high levels of M1 macrophages that are unable to switch to the M2 phenotype. This switch in an acute wound dampens inflammation and progresses the proliferative stage of repair but is missing from chronic wounds. For diabetic and chronic venous ulcers, immune cells are pre-primed prior to entering the wound by the low levels of chronic inflammation seen in these patients and factors such as AGEs and iron that alter macrophage phenotype. This pre-priming alters their ability to form M2 macrophages. In diabetes this is due to an inability to phagocytose spent neutrophils, in chronic venous ulcers the cause remains to be determined but may also be an inability to phagocytose. Future research will determine whether this is the case and how these cells might be altered to progress wound healing.
Author(s)
Katherine B Sahin
The Institute for Health and Biomedical Innovation, School of Biomedical Sciences, Faculty of Health, Queensland University of Technology,
Brisbane, QLD 4059, Australia
Mark Hesketh
The Institute for Health and Biomedical Innovation, School of Biomedical Sciences, Faculty of Health, Queensland University of Technology,
Brisbane, QLD 4059, Australia
Zoe E West
The Institute for Health and Biomedical Innovation, School of Biomedical Sciences, Faculty of Health, Queensland University of Technology,
Brisbane, QLD 4059, Australia
Rachael Z Murray*
The Institute for Health and Biomedical Innovation, School of Biomedical Sciences, Faculty of Health, Queensland University of Technology,
Brisbane, QLD 4059, Australia
Email rachael.murray@qut.edu.au
* Corresponding author
References
- Mahdavian Delavary B, van der Veer WM, van Egmond M, Niessen FB, Beelen RH. Macrophages in skin injury and repair. Immunobiology 2011;216(7):753–62.
- Abbade LP, Lastoria S, Rollo Hde A. Venous ulcer: clinical characteristics and risk factors. International J Dermatol 2011;50(4):405–411.
- Beitz JM, Goldberg E. The lived experience of having a chronic wound: a phenomenologic study. Medsurg Nurs 2005;14(1):51–62.
- Johannesson A, Larsson GU, Ramstrand N, Turkiewicz A, Wirehn AB, Atroshi I. Incidence of lower-limb amputation in the diabetic and nondiabetic general population: a 10-year population-based cohort study of initial unilateral and contralateral amputations and reamputations. Diabetes Care 2009;32(2):275–80.
- Graves N, Zheng H. Modelling the direct health care costs of chronic wounds in Australia. Wound Practice & Research 2014;22(1):20–33.
- Eming SA, Hammerschmidt M, Krieg T, Roers A. Interrelation of immunity and tissue repair or regeneration. Semin Cell Dev Biol 2009;20(5):517–27.
- Kim MH, Liu W, Borjesson DL et al. Dynamics of neutrophil infiltration during cutaneous wound healing and infection using fluorescence imaging. J Invest Dermatol 2008;128(7):1812–20.
- Rodero MP, Licata F, Poupel L, Hamon P, Khosrotehrani K, Combadiere C, Boissonnas A. In vivo imaging reveals a pioneer wave of monocyte recruitment into mouse skin wounds. PLoS One 2014;9(10):e108212.
- Schreml S, Szeimies R-M, Prantl L, Landthaler M, Babilas P. Wound healing in the 21st century. J Am Acad Dermatol 2010;63(5):866–881.
- Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol 2005;15(11): 599–607.
- Brancato SK, Albina JE. Wound macrophages as key regulators of repair: origin, phenotype, and function. Am J Pathol 2011;178(1):19–25.
- Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 2011;11(11):723–37.
- Rodero MP, Khosrotehrani K. Skin wound healing modulation by macrophages. Int J Clin Exp Pathol 2010;3(7):643–53.
- Daley JM, Brancato SK, Thomay AA, Reichner JS, Albina JE. The phenotype of murine wound macrophages. J Leukoc Biol 2010;87(1):59–67.
- Shook B, Xiao E, Kumamoto Y, Iwasaki A, Horsley V. CD301b+ Macrophages are essential for effective skin wound healing. J Invest Dermatol 2016;136(9):1885–91.
- Loots MA, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD, Middelkoop E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J Invest Dermatol 1998;111(5):850–7.
- Sindrilaru A, Peters T, Wieschalka S et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest 2011;121(3):985–97.
- Miao M, Niu Y, Xie T, Yuan B, Qing C, Lu S. Diabetes-impaired wound healing and altered macrophage activation: a possible pathophysiologic correlation. Wound Repair Regen 2012;20(2): 203–13.
- Okizaki S, Ito Y, Hosono K et al. Suppressed recruitment of alternatively activated macrophages reduces TGF-beta1 and impairs wound healing in streptozotocin-induced diabetic mice. Biomed Pharmacother 2015:70:317–25.
- Wallace HJ, Stacey MC. Levels of tumor necrosis factor-alpha (TNF-alpha) and soluble TNF receptors in chronic venous leg ulcers — correlations to healing status. J Invest Dermatol 1998;110(3):292–6.
- Patel S, Maheshwari A, Chandra A. Biomarkers for wound healing and their evaluation. J Wound Care 2016;25(1):46–55.
- Subramaniam K, Pech CM, Stacey MC, Wallace HJ. Induction of MMP-1, MMP-3 and TIMP-1 in normal dermal fibroblasts by chronic venous leg ulcer wound fluid*. Int Wound J 2008;5(1): 79–86.
- Ashcroft GS, Jeong MJ, Ashworth JJ et al. Tumor necrosis factor-alpha (TNF-alpha) is a therapeutic target for impaired cutaneous wound healing. Wound Repair Regen 2012;20(1):38–49.
- Streit M, Beleznay Z, Braathen LR. Topical application of the tumour necrosis factor-alpha antibody infliximab improves healing of chronic wounds. Int Wound J 2006;3(3):171–9.
- Mirza R, Koh TJ. Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine 2011;56(2): 256–64.
- Klinkert K, Whelan D, Clover AJP, Leblond AL, Kumar AHS, Caplice NM. Selective M2 macrophage depletion leads to prolonged inflammation in surgical wounds. Eur Surg Res 2017;58(3–4):109–120.
- Jetten N, Roumans N, Gijbels MJ et al. Wound administration of M2-polarized macrophages does not improve murine cutaneous healing responses. PLoS One 2014;9(7):e102994.
- Greenlee-Wacker MC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev 2016;273(1):357–70.
- Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol 2002;2(12):965–75.
- Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol 2005;6(12):1191–7.
- Filardy AA, Pires DR, Nunes MP, Takiya CM, Freire-de-Lima CG, Ribeiro-Gomes FL, DosReis GA. Proinflammatory clearance of apoptotic neutrophils induces an IL-12(low)IL-10(high) regulatory phenotype in macrophages. J Immunol 2010;185(4):2044–50.
- Das A, Ganesh K, Khanna S, Sen CK, Roy S. Engulfment of apoptotic cells by macrophages: a role of microRNA-21 in the resolution of wound inflammation. J Immunol 2014;192(3):1120–9.
- Khanna S, Biswas S, Shang Y et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One 2010;5(3):e9539.
- Liu BF, Miyata S, Kojima H, Uriuhara A, Kusunoki H, Suzuki K, Kasuga M. Low phagocytic activity of resident peritoneal macrophages in diabetic mice: relevance to the formation of advanced glycation end products. Diabetes 1999;48(10):2074–82.
- Guo Y, Lin C, Xu P, Wu S, Fu X, Xia W, Yao M. AGEs induced autophagy impairs cutaneous wound healing via stimulating macrophage polarization to M1 in diabetes. Sci Rep 2016;6:36416.
- Darby IA, Bisucci T, Hewitson TD, MacLellan DG. Apoptosis is increased in a model of diabetes-impaired wound healing in genetically diabetic mice. Int J Biochem Cell Biol 1997;29(1): 191–200.

