

Volume 26 Number 1
The multifactorial formation of chronic wounds
Rachael Z Murray, Zoe E West and William McGuiness
Keywords Diabetes, chronic wound, Inflammation, social factors, chronic venous insufficiency.
Abstract
In Australia, it is estimated that $2.85 billion is spent annually on wound care. As well as putting a significant strain on the health care system, these wounds decrease patient quality of life, and factors, such as immobility and isolation, can lead to depression. The real impact of these wounds is often masked by co-morbidities such as diabetes and cardiovascular disease. Current treatments available are not a ‘fix all’ solution, and many chronic wounds persist for months, even years, with a major limb amputation required every 30 seconds, amounting to 2500 amputations a day. Understanding how and why these wounds form, and why some heal, and others persist for years is necessary to improve the lives of these patients. Here we discuss, firstly, how co-morbidities alter the wound healing process at a cellular level to promote wound chronicity in patients and how therapies have developed from this information. Then we discuss the impact on the chronicity of wounds that factors, such as wound infection, the patient’s own social context and the health professional may have.
Introduction
Chronic wounds are seldom seen in otherwise healthy people and most patients have some sort of co-morbidity, such as diabetes or cardiovascular disease. This underlying disease pathology contributes to the formation and chronicity of these wounds, and our understanding of how this occurs has improved greatly over the last decade. Equally, factors other than the physiological have been identified as contributors to delayed healing. The social context, the psychological status of the patient and the competence of health care professionals combined with the physiological influences to produce a complex ‘pot-pourri’ of factors that warrant the attention of wound management practitioners.
Diabetic ulcers
Up to 25% of the 422 million diabetics worldwide will develop a chronic wound in their lifetime1. These patients have a 1% chance of having a limb amputated as a result of chronic wound progression, with one in four amputees requiring collateral amputation and/or re-amputation2,3. Understanding how these wounds form, and improving treatment is vital to reducing this statistic. A range of factors can influence the development of a chronic diabetic wound, including neuropathy, which often means injuries go undetected; hypoxia in the wound, due to insufficient perfusion and angiogenesis leads to reduced tissue oxygenation and nutrient supply, which can alter the immune response and damage tissue4; and high levels of glucose that can also alter immune cells and other functions in the wound.
In a healthy person, skin repair is a highly regulated process consisting of four consecutive phases: haemostasis, inflammation, proliferation and maturation that leads to wound closure (Figure 1)3. In the first phase, blood loss is limited by vasoconstriction and clot formation, and platelet degranulation releases cytokines and growth factors such as platelet-derived growth factor (PDGF) that initiate the inflammatory phase5. Neutrophils are recruited and their levels peak around 24–48 hours post injury. They remove dead cells, debris and pathogens in the wound, then go through a process of programmed cell death, known as apoptosis, to leave apoptotic bodies that are later efferocytosed (eaten) by macrophages5. Mediators released from these cells, and the injured environment, recruit monocytes from the circulation that differentiate into macrophages en route or at the site of injury. Macrophages remove the spent neutrophils, remaining debris and pathogens. They also regulate wound healing by secreting a plethora of cytokines, growth factors and other mediators that regulate the subsequent proliferation phase. The proliferation phase includes the formation of new blood vessels (angiogenesis), the deposition of extracellular matrix (ECM) to act as a scaffold for cells to populate the wound area, and the proliferation and migration of keratinocytes and fibroblasts to cover and repopulate the wound5. Wounds then mature with the remodelling ECM components5.
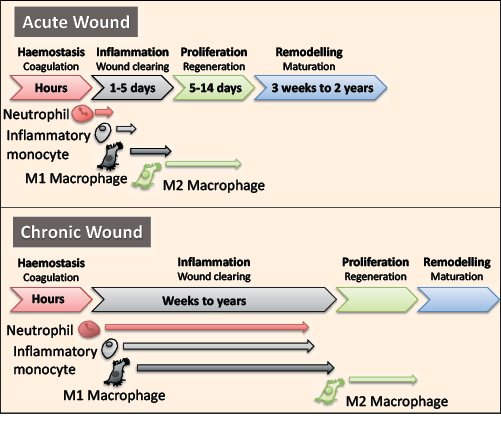
Figure 1: The inflammatory phase of wound healing is dysregulated in chronic wounds and can last from weeks to years. Wound healing consists of four overlapping phases: haemostasis, inflammation, proliferation and remodelling. The inflammatory phase is regulated by a number of innate immune cells including neutrophils, monocytes and macrophages. In a chronic wound, these cells are altered and persist in the wound to increase inflammation, which inhibits wound closure.
As with other chronic wounds, diabetic ulcers are typically characterised by a sustained and increased inflammatory phase (Figure 2), hypoxia, increased proteolytic activity that reduces the deposition of ECM components, and a reduced proliferative phase. Collectively, these often-interlinking factors contribute to the chronicity of wounds. It is now well established that the high levels of glucose in diabetic patients alters the immune system in numerous ways. This dysregulation directly increases and sustains the inflammatory phase, which prevents wound healing (Figure 1).
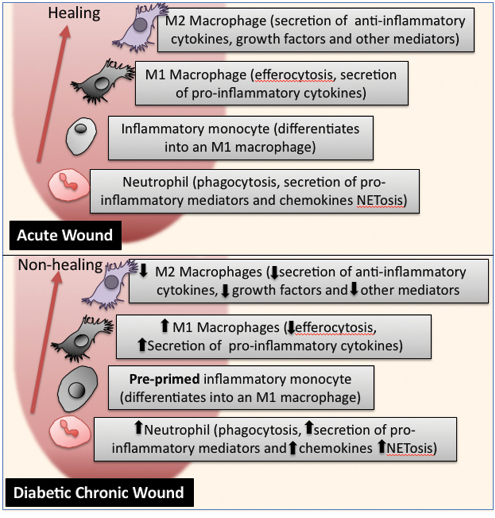
Figure 2: Diabetic chronic wounds contain innate immune cells with altered inflammatory functions.
Neutrophils, the first immune cells recruited, help clear wounds of pathogens. One way they do is this is through a process known as NETosis, where these cells release into the local environment neutrophil extracellular traps (NETs) formed from their decondensed chromatin. These NETs are lined with cytotoxic proteins and destructive enzymes that aid in the killing of pathogens. However, these NETs with their destructive proteins can impair wound healing and the levels in wounds need to be finely regulated so as not to cause extensive tissue damage and exacerbate inflammation. Neutrophils taken from both type 1 and 2 diabetic patients are pre-primed for NETosis6,7. Similarly, diabetic mice display a comparable pre-priming of neutrophils that in their wounds leads to increased NETosis6. This NETosis contributes to the delay in wound healing as reducing NETosis in the wound shortens the repair process6.
Neutrophils are not the only immune cells altered by high levels of glucose. Human monocytes can be divided up into different subsets based on surface CD14 and CD16 expression. Patients with type 2 diabetes have an increased number of CD14++CD16− inflammatory monocytes in their circulation8,9. These monocytes have enhanced inflammosome activation, which can lead to increased secretion of pro-inflammatory IL-1β and reactive oxygen species (ROS). ROS damages DNA and inhibit bacterial oxidative phosphorylation; however, excessive ROS in the local environment can increase inflammation and tissue damage. These same inflammatory monocytes that release pro-inflammatory cytokines such as TNF and IL-6 are also found in type I diabetic patients. Thus, glucose activation of these monocytes means they are pre-primed to increase inflammation when they arrive at an injury in the diabetic patients.
The macrophages that differentiate from these glucose-activated inflammatory monocytes have a defect in their ability to efferocytose dead neutrophils10. Efferocytosis by healthy (non-glucose activated) macrophages acts as a switch for the cell to change from a pro-inflammatory M1 macrophage to the anti-inflammatory M2 reparative macrophage that secretes anti-inflammatory cytokines, such as IL-1011. This switches off inflammation, leading to the conclusion of the inflammatory phase, allowing the later phases of the repair process to be completed. Mouse studies show that blocking the switch from M1 to M2 lengthens the inflammatory stage, suggesting defects in this switch might contribute to the excessive and prolonged inflammation found in chronic wounds12. In patients, approximately 80% of the cells found at the wound margin of diabetic ulcers are M1 pro-inflammatory macrophages, suggesting that this glucose-induced disruption of efferocytosis favours M1 pro-inflammatory macrophage phenotypes persisting in the wound13. Inflammatory macrophages secrete a range of proteins including pro-inflammatory cytokines, such as TNF and IL-6, as well as iNOS into the wound5. High levels of these proteins then act as chemoattractants and can cause damage to the local tissue, leading to more immune cell recruitment, thus increasing and perpetuating inflammation within the wound.
M1 macrophages also secrete enzymes, such as matrix metalloproteinases (MMPs), which help clear damaged ECM components and play a role in remodelling the ECM. The ECM acts a scaffold for new cells to populate the wound and its loss impedes wound closure. These proteases also cleave other proteins that regulate the repair process, such as TGFβ. Within diabetic wounds the levels of MMPs, and their endogenous inhibitors, are altered, with high MMP activity thought to keep the wounds in an inflammatory stage14,15. High levels are often a predictor of poor wound healing, with the wound fluid from patients that heal with best practice treatment showing a reduction in MMP9 as they heal, while MMP9 levels remain high in those that don’t heal14,15. These MMPs are secreted by M1 macrophages and neutrophils in the wound. Thus, the high level of M1 pro-inflammatory macrophages in diabetic ulcers leads to excessive damage, which, in turn, attracts more immune cells and the cycle of inflammation and damage continues.
In addition to the altered cells and molecules that are secreted in the wound environment these ulcers often exhibit high levels of hypoxia due to the disruptions in the vasculature around the wound reducing oxygen delivery. Hypoxia-inducible factor-1 (HIF-1) is a transcription factor responsible for the upregulation of genes, such as vascular endothelial growth factor (VEGF), that regulate the formation of new blood vessels (angiogenesis) to oxygenate the wound16. It also transcriptionally upregulates a plethora of genes that promote repair, such as transforming growth factor-β1 (TGF-β1), insulin-like growth factor 2 (IGF-2), type I collagen and fibronectin17. HIF-1 is normally continuously degraded but under conditions of hypoxia it is stabilised, active and promotes cell migration, cell survival, proliferation, growth factor release, and matrix synthesis18. Hyperglycaemia leads to HIF-1α destabilisation and functional repression, which inhibits HIF-1 signalling16. An imbalance in HIF-1 signalling results in impaired angiogenesis, with reduced levels of VEGF, angiopoietin 2, and fibroblast growth factor-2 (FGF-2) transcription, ultimately leading to delayed wound healing17. Endothelial progenitor cell motility is also reduced by impaired HIF-1 signalling, which contributes to the disrupted neovascularisation17. Fibroblasts extracted from diabetic patients do not upregulate VEGF in response to hypoxia due to glucose modifying the HIF-1α coactivator p30019. Diabetic mouse models show this prevents the upregulation of key repair genes such as heat shock protein 90 (HSP-90), VEGF-A and VEGF-R119. HIF-1 also regulates the expression of laminin-332 and so reduced HIF-1 signalling reduces its levels, which impairs the migration of keratinocytes and would impact on wound closure17.
Collectively, the alterations that diabetes imposes on the wound healing process promotes wound chronicity. Though the standard care for diabetic foot ulcers has improved immensely, approximately 10–15% of ulcers will remain active and 5–24% will lead to limb amputation, highlighting a need for target therapies to be developed/used in combination with good wound practice20. Our understanding of the key growth factors, cytokines and other mediators that regulate, or are secreted by cells in the wound environment, and impede wound healing has also greatly improved. This has led to the development of targeted therapies for chronic ulcers including anti-cytokine antibodies, autologous growth factors, recombinant growth factors and cell-based therapies21.
A number of different growth factors have been trialled on diabetic foot ulcers including PDGF, granulocyte colony-stimulating factor (GC-SF), transforming growth factor (TGF), fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF), insulin-like growth factor (IGF), and keratinocyte growth factor (KGF)22. A Cochrane systematic review of 28 randomised controlled trials (RTCs) using growth factors showed that there is insufficient evidence to recommend or refute the use of growth factors in treating diabetic foot ulcers22. This mainly seems due to the design of the clinical trials, inherent biases and lack of data in some areas such as amputations, mortality and quality of life to complete the picture.
Becaplermin (Regranex) is a recombinant human-PDGF approved in 1997 to treat diabetic foot ulcers that have a good enough blood supply and extend into the subcutaneous tissue or beyond. In RCTs it has been shown to accelerate diabetic wound closure, although it does not perform so well on venous ulcers21. It also has few limitations in that it is costly, requires frequent dressing changes that would interrupt standard limb compression therapy and there is an increased risk of malignancy when more than three tubes of Becaplermin are used on a patient. This could be altered by redesigning a delivery system that requires less growth factor and that could provide slow release so dressings are not changed as frequently. Recombinant bFGF has also been trialled on diabetic foot ulcers with variable results; one study showed no effect and another showing a significant effect21. Whether this is due to the grade of wound and concentration of recombinant bFGF used is unclear. Larger RCTs would need to be completed to answer these questions and as to whether they will help improve wound healing. Topical VEGF (Telbermin) underwent phase I and II trials but gave negative results and so has not progressed to the clinic. The variability in the results with growth factors suggest we may need greater understanding of the repair process to better target factors that might benefit wound healing and that better designed RCTs are needed when trialling these factors.
Venous and arterial ulcers
Often venous and arterial leg ulcers are classified together as they both result from insufficient blood flow, with venous leg ulcers being the most common ulcers23. Venous ulcers form in patients with sustained venous hypertension, attributed to chronic venous insufficiency (CVI) due to defective valves in the legs24. Arterial ulceration is less prevalent and patients have a reduced arterial supply to the lower extremities due to diseases such as atherosclerosis and vasculitis24. Of patients with chronic venous ulcers, 15–30% are classified as having mixed venous and arterial ulcers25. Venous and arterial ulcers, like diabetic ulcers, are often hypoxic, have high levels of inflammation with large numbers of M1 pro-inflammatory macrophages, and high levels of degradative enzymes26.
Chronic venous disease patients present with deep and superficial lower limb vein hypertension, which leads to oedema, the extravasation of macromolecules and red blood cells, leading to an excessive deposition of iron into the dermis. Their endothelial walls are altered due to inflammation and have increased levels of ICAM-1, an adhesion molecule involved in the recruitment of immune cells to tissue, which promotes inflammation27. Oedema and fibrosis impede oxygen diffusion and the supply of growth factors and nutrients into tissue and wounds. At a cellular level, chronic low levels of inflammation lead to both neutrophils and macrophages being activated prior to their entering a wound. Macrophages phagocytose the red blood cells that have leaked into tissue and so become iron-overloaded28. Mouse models of iron-overloaded wounds show that the immune cell composition at five days post injury is predominantly a pro-inflammatory M1 macrophage with an intermediate M2 macrophage phenotype that is still producing pro-inflammatory cytokines13. These wounds, like their diabetic counterparts, have high levels of some MMPs, including MMP2 and MMP9, that would contribute to the destruction of the ECM and prevent wound closure. All of these factors support delayed healing and contribute to the chronicity of wounds. With arterial diseases, a prolonged reduction in arterial supply can lead to tissue ischaemia and necrosis. Occlusions within the limb arteries leads to structural and functional changes in microcirculation and inflammation, which can similarly alter wound healing29.
Like with diabetic ulcers growth factors have also been trialled clinically to determine whether they might improve wound closure rates. For example, recombinant KGF (Repifermin) and recombinant PDGF (Becaplermin) have been tested but have provide mixed results21. However, high levels of the pro-inflammatory cytokine TNF have been shown, in multiple types of chronic wounds types including venous and arterial ulcers, to impede wound healing, making it a good target for therapy30,31. Pentoxifylline is a prostaglandin inhibitor known to have anti-TNFα properties. A meta-analysis of 12 randomised controlled studies showed that pentoxifylline is more effective than placebo (with or without compression), suggesting it might be of benefit to these patients32. The role of TNF in promoting inflammation in many diseases has also led to a number of good anti-TNF therapies that are in use in the clinic for other diseases, such as anti-TNF antibodies like infliximab and adalimumab30,31. In a pilot study, it was found that of 14 hard-to-heal ulcers (8 patients) given infliximab topically 5 healed, and 4 were reduced by more than 75% in size30,31. The wounds were of mixed aetiology, with CVI being the most prominent factor30,31. In a separate study, but this time treating with adalimumab, 4 of 5 patients with hard-to-heal venous leg ulcers showed an average reduction of 20.5% ± 6.4% wound size at 4 weeks30,31. These results suggest that targeting TNF might be beneficial, but they should be evaluated more thoroughly using well-designed RCTs.
Pressure ulcers
Unrelieved pressure, and/or the repeated shearing of tissue can result in microvascular ischaemia, causing damage to the underlying tissue and pressure ulcers33. When blood and lymphatic flow are occluded, waste products and proteins accumulate in the local tissue34. At a cellular level, much less is known about these wounds. Animal models of pressure show the up-regulation of genes involving apoptosis (programmed cell death), inflammation, oxidative stress, proteolysis and hypoxia35. Pro-inflammatory genes are upregulated at these sites of prolonged pressure, including IL-1β, and IL-6, and it is thought that, similar to other ulcer types, continuous local inflammation contributes to ulcer formation35. Stage III and IV patient chronic pressure ulcers show that, like other types of ulcers, these wounds have increased inflammatory cell infiltration36. Although macrophage phenotypes have not been looked at in these ulcers, the increased levels of IL-1β and TNF-α compared with acute wounds and normal skin suggest they are predominantly pro-inflammatory M1 macrophages36. Factors involved in processes such as angiogenesis for example, VEGF and bFGF, together with their receptors KDR and FGFR1, respectively, are also significantly decreased36. Interestingly, caspase-3 is elevated in these ulcers, suggesting that apoptosis (programmed cell death) is increased in these wounds, along with increased inflammation36. Whether the pressure injury is altering the immune cell phenotype in these wounds, similar to other wound types, is yet to be determined and it will be interesting to see whether similar changes occur as seen in the other types of chronic wounds.
Again, growth factors have been trialled on these chronic wounds. For example, recombinant bFGF has been use in at least two RCTs and found to be promising in terms of improving wound closure21. What all of the studies on growth factors and cytokines in the different types of ulcers tell us is that more work is needed to understand what is dysregulated in these wounds and how we might improve wound healing.
The contribution of wound infection to wound chronicity
One of the inevitable consequences of skin injury is the exposure to the skin microbiome. The wound environment is ideal for bacterial proliferation as it is warm, moist, provides nutrition from wound exudate and, in the case of diabetes, is high in glucose37. Our immune system is typically good at clearing pathogens from the wound, as is seen with the majority of acute wounds. However, in combination with other factors, such as reduced vascular supply and alterations in the host immune function, bacterial colonisation is more likely to occur and contribute to the chronicity of wounds.
Whilst colonisation is inevitable, infection is often dependent on a host response38. Chronic wound bacteria are 10 times more likely to form biofilms compared to acute wound bacteria39. These biofilms are harder to treat and are 500 times more likely to be resistant to antibiotics38. Due to the limited ability to culture anaerobic organisms, bacterial profiling has been variable within chronic wounds, although with methods like deep sequencing this is beginning to change. Despite the varying profiles, S. aureus is generally considered a universal coloniser of the different types of ulcers40. Interestingly, the skin microbiome of a diabetic patient’s feet has increased numbers of S. aureus, decreased numbers of Staphylococcus species, and increased bacterial diversity compared to non-diabetic control feet41. Whilst bacterial infection is not the sole factor in chronic wound development, it may tip already compromised healing into a state of chronicity. Infection promotes an influx of neutrophils and monocytes into wounds and this leads to an increase in secreted factors such as proteases, pro-inflammatory cytokines and ROS that could contribute to wound chronicity, depleting growth factor availability42,43.
The contribution of social context to wound chronicity
Alterations in cells and mediators such as cytokines and growth factors in the wound, along with infection, are not solely responsible for wound chronicity. Elements of a patient’s social context have been identified as contributors to delayed wound healing44-46. These are the occupation of the patient, access to health care services, nutritional habits and fiscal resources, to name a few. It is beyond the scope of this paper to discuss each, but a sample is provided. To help focus the discussion, venous ulceration and the accompanying CVI will be used as an example to highlight the relationship to chronic wound healing.
When considering occupations, CVI is believed to be higher in occupations that increase calf venous pressure. Principally, the relationship between long periods of standing and the prevalence and severity of the disease has been examined47-55. Population studies have demonstrated an association between standing and the prevalence of varicose veins. Tuchsen et al.54 followed 1.6 million Danish workers for a period of three years after their first hospital admission due to varicose veins of the lower extremities. They concluded that working in a standing position was associated with subsequent hospitalisation for varicose veins. Abramson et al.56 also found that prolonged standing contributed to the development of varicose veins.
In contrast, the study undertaken by Maffei et al.57 in Brazil found that working posture was not correlated with the incidence of varicose veins or venous ulceration. Likewise, Scott et al.51 who performed a multivariate analysis of patients with venous ulcers and a control group in the Boston area, found no significant association between occupation and periods of standing on the prevalence of venous ulceration. Clearly more research is needed, but the two schools of thought mandate consideration be given to occupation of the patient.
Links to dietary habits and chronic wounds have also been cited. Cleave58 initially proposed that a relationship existed between diet, specifically insufficient dietary fibre ('Western diet’) and the prevalence of varicose veins. He asserted that insufficient fibre led to constipation, which, in turn, increased intra-abdominal pressures. Burkitt et al.59,60 supported this position, citing the propensity of people from developing countries to develop similar prevalence rates to those of their adopted country. Fowkes et al.61 recorded subjects’ effort to open their bowels, their dietary fibre intake and transit times of faecal material within their bowel. Whilst they found an increased prevalence of trunk varices in men reporting that they had strained when opening their bowels, the findings did not demonstrate any significant association between fibre intake, faecal transit time and venous reflux.
In keeping with the influence of dietary habits on CVI, additional studies have examined associations between basal metabolic index and the prevalence of the disease. A number of authors argue that obesity is associated with CVI56,59,62-66. Danielsson et al.64, using a convenience sample, attended a Hawaiian hospital and investigated the impact that being overweight had on the severity of CVI. Subjects were classified as overweight (body mass index [BMI] >25 kg/m2) or obese (BMI >30 kg/m2) and were examined for clinical signs of CVI and the degree of venous reflux. They concluded that overweight patients were more likely to have skin changes and ulceration than patients with a BMI of less than 25 kg/m2. This was regardless of the amount of venous reflux present. Siedell et al.67 undertook a retrospective cohort study to examine the influence that being overweight had on chronic illness amongst patients attending four general practices in the Netherlands. They found that moderately overweight women were more likely to have varicose veins than normal weight women, and that obese women were three times more likely to have varicose veins. Abramson et al.56 examined a community in Western Jerusalem in an effort to identify factors associated with CVI. They found that the prevalence of varicose veins was 1.4 times higher amongst women than men and attributed this finding to the number of women in the study that were classified as overweight. As the problem of obesity continues to increase, the correlation between weight and delayed healing cannot be overlooked.
An association between social class and the duration and recurrence of leg ulcers is a common theme in the literature46,68-72. Many patients lived alone and had limited fiscal resources for purchasing care and the required nutrition to facilitate healing. Equally, they were unable to purchase preventative equipment such as compression hosiery, to reduce the risk of recurrence.
Phillips et al.71 found that 67% of participants stated that the presence of their leg ulcer impacted negatively on their financial situation. The main sources of expense identified were the cost of medical care, dressings and transportation. In over 30% of cases, these expenses were estimated to be between $101 and $1000 per annum. A further 8% of participants estimated out-of-pocket expenses to be greater than $1000 per annum.
A study conducted by Smith and McGuiness73 in Australia in 2006 identified that participants spent on average a total of $114 per month on the treatment and management of their ulcer, resulting in a personal cost of $1368 per annum. Sixty per cent of the costs incurred were for primary dressing products, with secondary dressing products (bandages, tapes), medications and transport accounting for the reminder of the expenditure. As the average age of participants in this study was 73.4 years, it could be assumed that the majority of participants in this study were financially supported by government pensions. Considering the average weekly income from aged pensions at that time was $206 (Australian Bureau of Statistics), the researchers concluded that it was likely that approximately 14% ($28) of participants’ total weekly income was being spent on the treatment and management of their leg ulcer. Kapp and Santamaria74 found similar financial impost on patient fiscal resources. Participants had spent on average A$2475 on wound products and A$121.82 in the most recent 28 days, representing 10% of their disposable income.
Time off work is commonly explored by researchers. Although a higher prevalence of ulceration is found in patients over the age of retirement, significant correlations were found between time lost from work, unemployment and impost on financial resources, and having a venous ulcer68,71,72. Inability to work or retain employment was associated with feelings of anger, resentment and associated depression.
It would be reasonable to expect that subsidisation of product would help to prevent wound chronicity. However, in an early study, Kapp at el.46 found no substantial improvement in the healing of venous leg ulcers when products were supplied. They did find that participants were significantly more likely to receive required medical footwear. A finding similar to the outcomes achieved by high-risk foot clinics for patients with diabetic foot ulcers75. More research is needed into subsidisation and the impact the financial stress has on a patient, but the contribution to delaying healing cannot be ignored.
The health professionals' contribution to chronic wounds
The ever-increasing evidence and growth of wound management expertise has improved the management of ‘at risk’ client cohorts, equitable access to services, and clinician naivety continues to contribute to the development of chronic wounds.
Patient dissatisfaction with health care workers is evident in qualitative studies. Hareendran et al.69 reported that 50% of the patients surveyed in their research indicated a disappointment with treatment. Ruckley72 suggested that this was related to the number of different health professionals patients encounter throughout the treatment. Because chronic wounds generally threatens neither life nor limb, patients are given less attention by busy professionals who ‘shunt’ them to other carers. The majority of their care is provided in the community by nurses and significant others70,76.
Specialist health care services are congregated within major city centres and often too few in number. Patients in rural or remote communities are often required to join a long waiting list and travel substantial distance to receive specialised care such as high-risk foot clinics or wound management centres. Telehealth is cited as a panacea to the tyranny of distance. The reality is that an already overburdened clinical specialist is still needed at one end of the exchange. Whilst the technology reduces the need to travel, it does not address the shortfall in experienced staff77.
The propensity to focus on product further places patients at risk of developing a chronic wound. Multiple suppliers in the market, combined with aggressive marketing campaigns has meant that clinicians and patients alike are confused about the ‘best product options’. As mentioned above, infection and the associated biofilms contribute to chronicity. Moving the focus from product selection to effective wound bed preparation may see reductions in time to heal.
Conclusion
Limiting the progression of acute wounds to chronic wounds requires a greater understanding of the healing process, new targeted therapies and the clinician to be vigilant on multiple fronts. Understanding and minimising the impact of underlying pathology, local infection, social constraints, and psychological outlook requires research, education and experience. This can only be achieved with financial support from government and private corporations. Researchers and health care professionals must continue to lobby for the allocation of such resources or be faced with an ever-increasing healing time and patient dissatisfaction.
Author(s)
Rachael Z Murray*
PhD, BSc
The Institute of Health and Biomedical Innovation, School of Biomedical Sciences, Faculty of Health, Queensland University of Technology, Brisbane, QLD 4059, Australia
Email rachael.murray@qut.edu.au
Zoe E West
BSc (MPhil student)
The Institute of Health and Biomedical Innovation, School of Biomedical Sciences, Faculty of Health, Queensland University of Technology, Brisbane, QLD, Australia
William McGuiness
PhD, FAWMA, RN, Dip T, BEd, MNS
The School of Nursing and Midwifery,
La Trobe University, VIC, Australia
* Corresponding author
References
- Singh N, Armstrong DG, Lipsky BA. Preventing foot ulcers in patients with diabetes. JAMA 2005;293(2):217–28.
- Sen CK, Gordillo GM, Roy S et al. Human skin wounds: A major and snowballing threat to public health and the economy. Wound Repair Regen 2009;17(6):763–71.
- Sahin KB, Hesketh M, West ZE, Murray RZ. Altered macrophage phenotypes impair wound healing. Wound Practice & Research 2017;25(4):166–71.
- Guo S, DiPietro LA. Factors affecting wound healing. J Dent Res 2010;89(3):219–29.
- Hesketh M, Sahin KB, West ZE, Murray RZ. Macrophage phenotypes regulate scar formation and chronic wound healing. Int J Mol Sci 2017;18(7).
- Wong SL, Demers M, Martinod K et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med 2015;21(7):815–9.
- Menegazzo L, Ciciliot S, Poncina N et al. NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol 2015;52(3):497–503.
- Giulietti A, van Etten E, Overbergh L, Stoffels K, Bouillon R, Mathieu C. Monocytes from type 2 diabetic patients have a pro-inflammatory profile. 1,25-Dihydroxyvitamin D(3) works as anti-inflammatory. Diabetes Res Clin Pract 2007;77(1):47–57.
- Yang M, Gan H, Shen Q, Tang W, Du X, Chen D. Proinflammatory CD14+CD16+ monocytes are associated with microinflammation in patients with type 2 diabetes mellitus and diabetic nephropathy uremia. Inflammation 2012;35(1):388–96.
- Khanna S, Biswas S, Shang Y et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One 2010;5(3):e9539.
- Filardy AA, Pires DR, Nunes MP et al. Proinflammatory clearance of apoptotic neutrophils induces an IL-12(low)IL-10(high) regulatory phenotype in macrophages. J Immunol 2010;185(4):2044–50.
- Klinkert K, Whelan D, Clover AJP, Leblond AL, Kumar AHS, Caplice NM. Selective M2 Macrophage Depletion Leads to Prolonged Inflammation in Surgical Wounds. Eur Surg Res 2017;58(3–4):109–20.
- Sindrilaru A, Peters T, Wieschalka S et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest 2011;121(3):985–97.
- Muller M, Trocme C, Lardy B, Morel F, Halimi S, Benhamou PY. Matrix metalloproteinases and diabetic foot ulcers: the ratio of MMP-1 to TIMP-1 is a predictor of wound healing. Diabet Med 2008;25(4):419–26.
- Liu Y, Min D, Bolton T et al. Increased matrix metalloproteinase-9 predicts poor wound healing in diabetic foot ulcers. Diabetes Care 2009;32(1):117–9.
- Catrina SB, Zheng X. Disturbed hypoxic responses as a pathogenic mechanism of diabetic foot ulcers. Diabetes Metab Res Rev 2016;32(Suppl 1):179–85.
- Sergiu-Bogdan C, Xiaowei Z. Distributed hypoxic responses as a pathogenic mechanism of diabetic foot ulcers. Diabetes Metab Res Rev 2016(32):179–85.
- Hong WX, Hu MS, Esquivel M et al. The role of hypoxia-inducible factor in wound healing. Adv Wound Care (New Rochelle) 2014;3(5):390–9.
- Thangarajah H, Yao D, Chang EI et al. The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. PNAS 2009;106(32):13505–10.
- Alexiadou K, Doupis J. Management of diabetic foot ulcers. Diabetes Ther 2012;3(1):4.
- Barrientos S, Brem H, Stojadinovic O, Tomic-Canic M. Clinical application of growth factors and cytokines in wound healing. Wound Repair Regen 2014;22(5):569–78.
- Marti-Carvajal AJ, Gluud C, Nicola S et al. Growth factors for treating diabetic foot ulcers. Cochrane Database Syst Rev 2015(10):CD008548.
- Vasudevan B. Venous leg ulcers: pathophysiology and classification. Indian Dermatol Online J 2014;5(3):366–70.
- Grey JE, Harding KG. Venous and arterial leg ulcers. BMJ 2006;332(7537):347–50.
- De Caridi G, Massara M, Stilo F et al. Effectiveness of prostaglandin E1 in patients with mixed arterial and venous ulcers of the lower limbs. Int Wound J 2014;13(5):625–9.
- Herber OR, Schnepp W, Rieger M. A systematic review on the impact of leg ulceration on patients’ quality of life. Health Qual Life Outcomes 2007;5(44).
- Chi WY, Raffetto JD. Venous leg ulceration pathophysiology and evidence-based treatment. Vasc Med 2015;20(2):168–81.
- Crawford JM, Lal BK, Duran WN, Pappas PJ. Pathophysiology of venous ulceration. J Vasc Surg Venous Lymphat Disord 2017;5(4):596–605.
- Weir G, Smart H, van Marie J, Cronje FJ. Arterial Disease Ulcers, Part 1: Clinical Diagnosis and Investigation. Adv Skin Wound Care 2014;27(9):421–8.
- Fox JD, Baquerizo-Nole KL, Keegan BR et al. Adalimumab treatment leads to reduction of tissue tumor necrosis factor-alpha correlated with venous leg ulcer improvement: a pilot study. Int Wound J 2016;13(5):963–6.
- Streit M, Beleznay Z, Braathen LR. Topical application of the tumour necrosis factor-alpha antibody infliximab improves healing of chronic wounds. Int Wound J 2006;3(3):171–9.
- Jull A, Arroll B, Parag V, Waters J. Pentoxifylline for treating venous leg ulcers. Cochrane Database Syst Rev 2007(3):CD001733.
- Smith N, Overland J, Greenwood J. Local management of deep cavity wounds — current and emerging therapies. Dove Press 2015;2015(2):159–70.
- Bhattacharya S, Mishra RK. Pressure ulcers: Current understanding and newer modalities of treatment. Indian J Plast Surg 2015;48(1):4–16.
- Kursoe T, Hashimoto M, Ozawa J, Kawanmata. Analysis of gene expression in experimental pressure ulcers in the rat with special reference to inflammatory cytokines. PLOS One 2015;Epub 1371.
- Jiang L, Dai Y, Cui F et al. Expression of cytokines, growth factors and apoptosis-related signal molecules in chronic pressure ulcer wounds healing. Spinal Cord 2014;52:145–51.
- Young L. Identifying infection in chronic wounds. Wound Practice & Research 2012;20(1):38–44.
- Siddiqui A, Bernstein J. Chronic wound infection: Facts and controversies. Clin Dermatol 2010;28(5):519–26.
- James GA, Swogger E, Wolcott R et al. Biofilms in chronic wounds. Wound Repair Regen 2007;16(1):37–44.
- Kirketerp-Møller K, Jensen PØ, Fazli M et al. Distribution, Organization, and Ecology of Bacteria in Chronic Wounds. J Clin Microbiol 2008;46(8):2717–22.
- Redel H, Gao Z, Li H et al. Quantitation and composition of cutaneous microbiota in diabetic and nondiabetic men. J Infect Dis 2013;207(7):1105–14.
- Zhao R, Liang H, Clarke E, Jackson C, Xue M. Inflammation in Chronic Wounds. Int J Mol Sci 2016;17(12):2085.
- Edwards R, Harding KG. Bacteria and wound healing. Curr Opin Infect Dis 2004;17(2):91–6.
- Parker CN, Finlayson KJ, Shuter P, Edwards HE. Risk factors for delayed healing in venous leg ulcers: a review of the literature. Int J Clin Pract 2015;69(9):967–77.
- Järbrink K, Ni G, Sönnergren H et al. The humanistic and economic burden of chronic wounds: a protocol for a systematic review. Syst Rev 2017;6(15).
- Kapp S, Miller C, Elder K. The impact of providing product funding for compression bandaging and medical footwear on compression use, wound healing and quality of life. Int Wound J 2012;9(5):494–504.
- Kroeger K, Ose C, Rudofsky G, Roesener J, Hirche H. Risk factors for varicose veins. Int J Angiol 2004;23(1):29–34.
- Lacroix P, Aboyans V, Preux PM, Houles MB, Laskar M. Epidemiology of venous insufficiency in an occupational population. Int J Angiol 2003;22(2):172–6.
- Pinto A, Corrao S, Galati D et al. Sulodexide versus calcium heparin in the medium-term treatment of deep vein thrombosis of the lower limbs. Angiology 1997;48(9):805–11.
- Sadick NS. Predisposing factors of varicose and telangiectatic leg veins. J Dermatol Surg Oncol 1992;18(10):883–6.
- Scott TE, LaMorte WW, Gorin DR, Menzoian JO. Risk factors for chronic venous insufficiency: a dual case-control study. J Vasc Surg 1995;22(5):622–8.
- Sisto T, Reunanen A, Laurikka J et al. Prevalence and risk factors of varicose veins in lower extremities: mini-Finland health survey. Eur J Surg 1995;161(6):405–14.
- Stvrtinova V, Kolesar J, Wimmer G. Prevalence of varicose veins of the lower limbs in the women working at a department store. International Angiology 1991;10(1):2–5.
- Tuchsen F, Krause N, Hannerz H, Burr H, Kristensen TS. Standing at work and varicose veins. Scand J Work Environ Health 2000;26(5):414–20.
- Brand FN, Dannenberg AL, Abbott RD, Kannel WB. The epidemiology of varicose veins: the Framingham Study. Am J Prev Med 1988;4(2):96–101.
- Abramson JH, Hopp C, Epstein LM. The epidemiology of varicose veins. A survey in western Jerusalem. J Epidemiol Community Health 1981;35(3):213–7.
- Maffei FH, Magaldi C, Pinho SZ et al. Varicose veins and chronic venous insufficiency in Brazil: prevalence among 1755 inhabitants of a country town. Int J Epidemiol 1986;15(2):210–7.
- Cleave TL. Varicose veins, nature’s error or man’s? Some implications of the Darwinian theory. Lancet 1959;2:172–5.
- Burkitt DP. Some diseases characteristic of modern Western civilization. Br Med J 1973;1(5848):274–8.
- Burkitt DP. Varicose veins in developing countries. Lancet 1976;1(7982):472.
- Fowkes FG, Lee AJ, Evans CJ, Allan PL, Bradbury AW, Ruckley CV. Lifestyle risk factors for lower limb venous reflux in the general population: Edinburgh Vein Study. Int J Epidemiol 2001;30(4):846–52.
- Beebe-Dimmer JL, Pfeifer JR, Engle JS, Schottenfeld D. The epidemiology of chronic venous insufficiency and varicose veins. Ann Epidemiol 2005;15(3):175–84.
- Carpentier PH, Maricq HR, Biro C, Poncot-Makinen CO, Franco A. Prevalence, risk factors, and clinical patterns of chronic venous disorders of lower limbs: a population-based study in France. J Vasc Surg 2004;40(4):650–9.
- Danielsson G, Eklof B, Grandinetti A, Kistner RL. The influence of obesity on chronic venous disease. J Vasc Endovasc Surg 2002;36(4):271–6.
- De Backer G. Epidemiology of chronic venous insufficiency. Angiology 1997;48(7):569–76.
- Jawien A. The influence of environmental factors in chronic venous insufficiency. Angiology 2003;54 Suppl 1:S19–31.
- Seidell JC, Bakx KC, Deurenberg P, van den Hoogen HJ, Hautvast JG, Stijnen T. Overweight and chronic illness –– a retrospective cohort study, with a follow-up of 6–17 years, in men and women of initially 20–50 years of age. J Chronic Dis 1986;39(8):585–93.
- Farias Alves Yamada B, Conceicao de Gouveia Santos V. Quality of Life of Individuals with Chronic Venous Ulcers. Wounds: A Compendium of Clinical Research & Practice [Internet]. 2005 30/110/2007; 17(7):[178–89 pp.].
- Hareendran A, Bradbury A, Budd J et al. Measuring the impact of venous leg ulcers on quality of life. J Wound Care 2005;14(2):53–7.
- Moffatt CJ, Franks PJ, Doherty DC, Smithdale R, Martin R. Sociodemographic factors in chronic leg ulceration. Br J Dermatol 2006;155(2):307–12.
- Phillips T, Stanton B, Provan A, Lew R. A study of the impact of leg ulcers on quality of life: financial, social, and psychologic implications. J Am Acad Dermatol 1994;31(1):49–53.
- Ruckley CV. Socioeconomic impact of chronic venous insufficiency and leg ulcers. Angiology 1997;48(1):67–9.
- Smith E, McGuiness B. Managing venous leg ulcers in the community: Financial cost to sufferers. [Honours thesis]. In press, 2006.
- Kapp S, Santamaria N. The financial and quality-of-life cost to patients living with a chronic wound in the community. Int Wound J 2017;14(6):1108–19.
- Holstein P, Ellitsgaard N, Olsen BB, Ellitsgaard V. Decreasing incidence of major amputations in people with diabetes. Diabetologia 2000;43(7):844–7.
- Rayner R. The role of nurse-led clinics in the management of chronic leg wound. Primary Intention 2006;14(4):162–7.
- Barrett M, Larson A, Carville K, Ellis I. Challenges faced in implementation of a telehealth enabled chronic wound care system. Rural Remote Health 2009;9(3):1154.

