

Volume 32 Number 1
The negative impact of medications on wound healing
Giselle Bennett, Julie Abbott, Geoff Sussman
Keywords wound healing, Adverse effects, drug-related, medication
For referencing Bennett G, Abbott J and Sussman G. The negative impact of medications on wound healing. Wound Practice and Research. 2024;32(1):17-24.
DOI
10.33235/wpr.32.1.17-24
Submitted 16 February 2024
Accepted 20 February 2024
Abstract
Chronic wounds can lead to amputations and significant decreases in quality of life. Many commonly used medications are known to cause ulcers or perpetuate chronic wounds. A variety of medication classes can impair wound healing through affecting cells within the skin, metabolism, immune cell function, angiogenesis and coagulation. This review aims to highlight the main types of drugs which negatively impact wound healing. Cancer treatments, non-steroidal anti-inflammatory drugs (NSAIDs), anticoagulants, immunosuppressants, and some antibiotics are all risk factors for cutaneous adverse effects. Identifying drug-induced impaired wound healing is important to counsel patients and their medical practitioners on weighing up the benefits and risks of these medications.
Introduction
Medication is an essential part of disease management and plays a vital role in both treatment of acute and chronic diseases. When a patient has an acute or chronic wound, the use of any medication which may impact on or delay wound healing must be considered. It is essential to obtain a full medication history including prescribed, ‘over-the-counter’ and complementary products being taken by the patient including oral, injected, topical or inhaled formulations. Risks and benefits must be recognised and weighed up by clinicians and patients to make informed decisions about whether to cease or dose-reduce these medications that can delay wound healing.
Impact of medication on wound healing
Medication use in patients for the management of their chronic diseases plays an important role in either the stimulation or inhibition of wound healing. Pharmaceuticals are used both directly and indirectly in wound management practice. Drugs are applied topically and used systemically as part of wound management for infection, pain management and sometimes immunosuppression for autoimmune aetiologies. Medications interfere with specific phases of wound healing and will affect cells, pathways, growth factors, cytokines, and other important components of the wound healing cascade. In addition, some drugs will, as part of their side-effects, reduce blood flow, blood cells and organ functions critical to wound healing.
Antineoplastic drugs
Chemotherapy can have wound healing complications that can lead to devastating consequences including loss of limb function. This risk is particularly concerning when cancer surgery is performed in conjunction with chemotherapy due to potential surgical wound complications. Some intravenous chemotherapy drugs can induce vein irritation which can result in non-healing necrotic ulcers.1
There are a variety of mechanisms of impaired wound healing due to chemotherapy, such as inhibiting cellular metabolism, cell division or angiogenesis. Many chemotherapeutics disrupt DNA replication, transcription, and translation. In wounds they impede cell migration, reduce extracellular matrix production, and inhibit fibroblast proliferation.1‑3 Consequences of these mechanisms include apoptosis, cell cycle arrest, senescence, mitotic catastrophe, inflammatory responses and fibrosis.4 Chemotherapy can severely impair wound healing through profound immunosuppression. As such, wounds may become infected due to decreased neutrophil and macrophage activity delaying removal of dead tissue and foreign bodies from the wound.5
Chemotherapy drugs purposefully target rapidly dividing cancer cells; however, they can also affect susceptible proliferating cells involved in skin wound healing. Some conditions such as excessive dry skin from chemotherapeutic agents can be complicated by cracks and open wounds and infections. One serious complication is hand-foot syndrome which is caused by 5-fluorouracil derivatives such as capecitabine. It is characterized by numbness and paraesthesia in the hands and feet that can quickly progress to serious ulceration and blisters.6
Chemotherapy causes apoptosis or dysplasia of rapidly dividing cell types which include keratinocytes, hair matrix keratinocytes (which causes alopecia), fibroblasts and melanocytes.4,7,8 Cyclophosphamide is associated with a higher risk of severe keratinocyte dysplasia.8 Cyclophosphamide reduces vasodilatation and subsequent neovascularisation during the proliferative phase of wound healing. Furthermore, chemotherapy has been shown to impair multiple stem cell types in skin including mesenchymal stromal stem cells (MSCs), epidermal stem cells (EPSCs), and hair follicle stem cells (HFSCs).1,9,10
Vesicants
Some chemotherapy drugs are vesicants which when extravasated can inflict permanent tissue damage. Symptoms of vesicant extravasation include erythema and pain that can progress to blistering, desquamation, necrosis, eschar formation and ulceration. The classes of chemotherapy that are vesicants include DNA-binding drugs (mustard gas derivatives, anthracyclines, dactinomycin), and non-DNA-binding drugs (vinca alkaloids, alkylators and taxanes).11 The DNA-binding agents remain bound to nucleic acids even after cell death and subsequently, when endocytosed by adjacent cells will lead to a repeating cycle of cell death that enlarges the wound. Non-DNA binding agents are metabolised and do not perpetuate in this way, with less breakdowns in skin integrity.12 Prompt cessation of the infusion when extravasation is recognised is key to limiting the tissue damage.
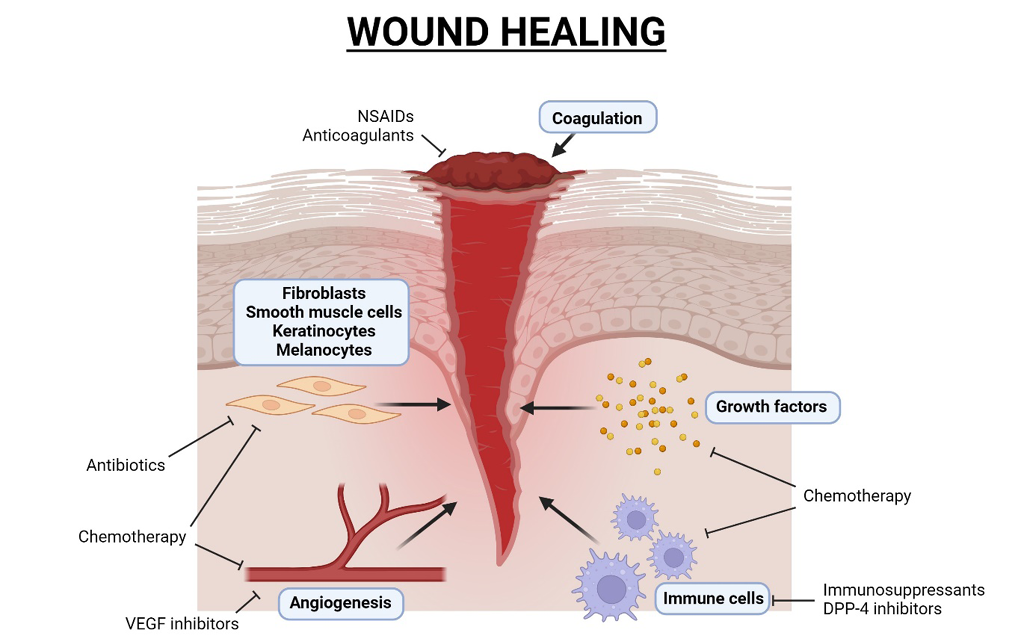
Figure 1. Diagram of wound healing and medications that impair wound healing processes. Created with Biorender.com
Hydroxyurea
Hydroxyurea is a cytostatic agent that inhibits DNA synthesis as a ribonucleotide reductase inhibitor. Hydroxyurea is one of the most widely publicised medications to cause leg ulcers. It is used for treatment of myeloproliferative disorders. Sirieix et al13 reported a retrospective series of 41 patients who developed leg ulcers during long-term use of hydroxyurea. Most cases (80%) had complete recovery after cessation of hydroxyurea in a mean of 3 months (range of 1–24 months). In the remaining cases, the ulcers had a reduction in size after discontinuation. Many patients had multiple ulcers, and the ulcers were located near the malleoli or on the calf and foot. For 70% of these patients, it was their first episode of leg ulcers. All ulcers were painful and 25% of cases were necrotic. A typical histopathology involves dermal fibrosis, scar tissue and epidermal atrophy. The mechanism for these ulcers is direct cytological damage due to hydroxyurea. Recommencing hydroxyurea is associated with recurrence of ulceration which confirms hydroxyurea as the cause.14 In the most serious cases of hydroxyurea ulceration, leg amputation may be required.15
Chemotherapy affecting growth factors and angiogenesis
Chemotherapy blocks the synthesis of growth factors subsequently causing decreased cell migration, decreased proliferation and reduced angiogenesis.1
Capecitabine and oxaliplatin have been shown to cause reduction in measured levels of growth factors including epidermal growth factor (EGF), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF).16 These growth factors are important for angiogenesis, hence, the anti-angiogenic properties of chemotherapy may be a key cause for impaired wound healing in these patients.1
VEGF plays multiple roles in wound healing: recruitment of monocytes, macrophages, fibroblasts and endothelial cells, increased vascular permeability, and deposition of collagen.17 The monoclonal antibody VEGF inhibitor bevacizumab works as a treatment for colon cancers by inhibiting new vessel formation, altering vascular function and decreasing tumour blood flow.18 However, angiogenesis is important for wound healing, and reports of impaired wound healing have occurred in patients on bevacizumab.19,20 This risk of surgical wound complications can be mitigated by administering bevacizumab 28–60 days after surgery due to its 20 day half-life.19 Additionally, VEGF inhibitors may be withheld during wound healing. Withholding bevacizumab was implemented to manage a patient with a large necrotic surgical wound and subsequent colostomy dehiscence which developed after bevacizumab and chemotherapy administration.21
Targeted cancer treatments
As more directed cancer treatment drugs become more widely available, there have been reports of skin ulcers with targeted cancer treatments as contributing causes. Targeted cancer treatments including tyrosine kinase inhibitors and VEGF inhibitors have been implicated in impaired wound healing.22,23
In 2016, there were two case reports of tyrosine kinase inhibitor induced foot wounds with sunitinib and nilotinib, however, the case treated with nilotinib had a confounding factor of premorbid peripheral arterial disease.24 Tyrosine kinase inhibitor use was independently associated with higher risk of post-operative complications after nephrectomy for stage IV renal cell carcinoma. Complications were broadly defined and included both vascular and wound healing complications.25 In 2022, Matsuo reported a case of pharyngocutaneous fistula developing after pharyngeal surgery for cancer where imatinib, a platelet-derived growth factor receptor alpha inhibitor was suspected to be implicated as a causative factor.23
Non-steroidal anti-inflammatory drugs (NSAIDS)
NSAIDs are members of a therapeutic drug class which reduces pain, decreases inflammation, decreases fever, and prevents blood clotting. There are two general types of NSAIDs available: non-selective, and COX-2 selective. COX-2 selective drugs include celecoxib, etoricoxib, and meloxicam. Most NSAIDs are non-selective and inhibit the activity of both COX-1 and COX-2; examples include naproxen, ibuprofen, diclofenac, indomethacin. These NSAIDs, while reducing inflammation, also inhibit platelet aggregation and increase the risk of gastrointestinal ulcers and bleeds. Their mode of action is to inhibit inflammatory response and acid mucopolysaccharide synthesis in wounds, inhibit inflammatory mediators derived from arachidonic acid metabolism and platelet aggregation. NSAIDs inhibit the production of prostaglandins (PGs), which may be the likely mechanism through which NSAIDs impart their deleterious effects on bone healing. By inhibiting the COX enzymes and the subsequent PG production, NSAIDs not only achieve their desired anti-inflammatory effects but also inhibit the increased production of PGs that is necessary for bone healing to occur. Additionally, the inhibition of COX-1 can increase the local ischemia and hypoxia associated with chronic venous ulcers.26
A study found reduced ligament repair and strength in surgically incised medial collateral ligament in 50 rats treated post operatively with a COX-2 inhibitor when compared with the non-treated rats.27 The inhibition of migration and proliferation of tendon cells by NSAIDs has also been demonstrated by Tsai et al.28,29 In animals treated with indomethacin or parecoxib, the Achilles tendon has decreased tensile strength compared to the control groups.30,31. Timing of NSAID use is important perioperatively, as the use of indomethacin for the first 7 days after surgery has been shown to contribute to the deterioration of healing compared to treatment after 7 days. Indomethacin has also been shown to have negative effects on the proliferation of human tenocyte cultures.32
Anticoagulants
Warfarin
Warfarin has a rare but well-known adverse effect of skin necrosis occurring in an estimated 0.01-0.1% of patients with over 300 cases reported.33,34 Cases of gangrenous skin necrosis attributed to anticoagulant therapy has been described since the 1950s.35,36 The onset of warfarin-induced skin necrosis (WISN) often occurs between day 1–10, most often between day 3–6.37 The disease is usually unilateral, however, 30% of cases have bilateral lesions.34,38–41 The condition has a female predominance, with a 4-fold to 9-fold risk compared to men, and in females the lesions more commonly affect the breast.33,34,41-43 WISN predominantly affects women treated for acute thromboembolic disease and has not been reported when warfarin is used for atrial fibrillation.34 The lesions in WISN typically localise in an areas of skin with abundant subcutaneous fat such as the breast, buttock, abdomen or thigh, and has a weak association with obesity.34,38,41,43,44 They begin as an area of erythema or petechiae that progress to haemorrhagic bullae, and subsequent necrotic eschar. Surgical debridement is required in 50% of cases.41,42,45 Nalbandian et al37 proposed that the pathophysiology of WISN begins with a direct toxic effect causing vascular dilatation at the arteriocapillary loop of the dermis, specifically at the junction of precapillary arteriole and capillary. This correlates clinically to the initial erythematous flush. However, lesions are not related to drug dose or duration. Next, damaged capillaries rupturing may correlate to the appearance of petechiae. Ecchymosis results from coalescent haemorrhages, as a consequence of the anticoagulant effect. The necrotic stage develops after venules thrombose distal to the dermo-vascular loop due to stasis.37 The pathogenesis appears to involve a combination of changes in haemostasis regulation and local vascular changes. WISN is associated with low levels of protein C, and there have also been reports of WISN in patients with low levels of free protein S. When warfarin is commenced, due to the inhibition of vitamin K-dependent factor production, there is a faster reduction in protein C and factor VII levels due to its shorter half-life compared to the other coagulation factors (factor IX, factor X, protein S and factor II).43, 44 This leads to a transient hypercoagulability until levels of procoagulant factors sufficiently decrease. Protein C and S deficiencies also cause a hypercoagulable state, therefore, the combination of protein C or S deficiencies with warfarin initiation is theorised to exacerbate thrombosis in microcirculation of the skin leading to skin necrosis.33,44 Protein C and total and free protein S levels should be measured in patients consistent with hypercoagulable state. Preventing WISN in people with hypercoagulable states may also involve heparin therapy as bridging anticoagulation to counteract the temporary initial hypercoagulable state with warfarin treatment.39 Ceasing warfarin and changing to a direct oral anticoagulant is the usual management to manage the anticoagulation requirements of these patients, meanwhile, treatment of the skin necrosis often requires surgical intervention.
Heparin
Heparins are polysaccharides that inactivate many coagulation factors and are widely used to treat and prevent thrombotic disorders. Heparin can cause adverse effects related to its anticoagulant effect such as purpura and haematomas either at the site of the injection or elsewhere.46 Another adverse effect is heparin-induced thrombocytopaenia type II (HIT II) which may lead to arterial and venous thrombosis and subsequent skin necrosis. The mechanism of HIT is the formation of autoantibodies against heparin platelet factor 4 complexes. The onset of HIT II is usually within the first 10–14 days of treatment, and the prevalence is approximately 0.1–2% of patients treated with heparin.46
Direct Oral Anticoagulants (DOACs)
DOACs are widely used and there have been reports of rare serious cutaneous side effects.47 There have been two case reports of dabigatran causing leukocytoclastic vasculitis.48,49 In 2012, Tsoumpris et al50 also described a case of toxic epidermal necrolysis (TEN), although the precipitating drug could have been either iron protein succinylate or dabigatran, or an interaction between the two drugs. There has been one case report of rivaroxaban-induced drug reaction with eosinophilia and systemic symptoms (DRESS) and liver injury in 2015.51 In 2021, Pansuriya52 described a case report of apixaban causing skin necrosis, which subsequently improved after discontinuing apixaban and changing to low-molecular-weight heparin. It is unknown whether warfarin and heparin related skin toxicities share the same pathophysiology as DOAC-skin toxicities.
Immunosuppressants
Methotrexate
Methotrexate, a folic acid antagonist inhibits DNA synthesis and cell replication by competitive inhibition of dihydrofolate reductase impairing folic acid conversion to folinic acid, providing immunosuppressive and anti-inflammatory actions.53 Thymidine synthetase activity is reduced resulting in deficient conversion of 2-deoxyuridinylate to thymidine, which subsequently partially blocks DNA and RNA synthesis.53 Methotrexate inhibits IL-1 and decreases IL-6 synthesis.53 It is indicated for the treatment of rheumatoid arthritis, ectopic pregnancy, certain cancers and psoriasis. Adverse effects include rare skin reactions, skin ulceration, Stevens-Johnson syndrome, and toxic epidermal necrolysis. Some people receiving high doses of methotrexate experience skin erosions.54 Biopsies of patients treated with high dose methotrexate show a variety of keratinocyte dystrophies.54 Leucovorin can be used to minimise risk of adverse effects with methotrexate in cancer patients.
Azathioprine
Azathioprine (AZA) is a purine antimetabolite which is metabolised via 6-mercaptopurine to its active metabolite 6-thioguanine nucleotide (6-TGN). Because AZA suppresses inosinic acid and purine synthesis, it interferes with B and T lymphocyte proliferation, T cell mediated immune reactions (decreased IL-2 secretion) and antibody responses.55 T lymphocytes play an important role in wound healing especially during the inflammatory phase. AZA is indicated for organ transplant rejection prevention, rheumatoid arthritis, inflammatory bowel disease and systemic lupus erythematosus. Adverse effects include Stevens-Johnson syndrome and toxic epidermal necrolysis.
Leflunomide
Leflunomide inhibits pyrimidine synthesis through selective dihydroorotate dehydrogenase and tyrosine kinase enzyme block-aid.56 Active metabolite teriflunomide acts on rapidly dividing cells and lymphocytes inhibiting their effects.57 Leflunomide’s toxic effect on epidermal cell lines impairs ulcer healing and arrests the production of epidermal growth factor.56 Skin ulceration with an incidence rate of 1–3% from post-marketing surveillance is listed in the American Hospital Formulary Service.56 It exhibits immunosuppressive, immunomodulating and antiproliferative properties. It is indicated for rheumatoid arthritis and psoriatic arthritis. Adverse effects include Stevens-Johnson syndrome and toxic epidermal necrolysis. Teriflunomide has an elimination half-life of approximately 2–4 weeks, therefore, its effects on wound healing can remain post cessation.58
Ciclosporin
Ciclosporin is a calcineurin inhibitor, forming a complex with cyclophilin inhibiting calcineurin action in activated T cells.55 Calcineurin inhibition prevents cytokine gene expression, reducing IL-2, IL-4 and TNF-α production and consequent T cell proliferation and differentiation.55 It is indicated for transplant rejection prevention, psoriasis, severe rheumatoid arthritis and nephrotic syndrome.
Mammalian target of rapamycin (mTOR) inhibitors
Sirolimus is a first-generation mTOR inhibitor used for prevention of transplant rejection. Impaired wound healing after surgery has been reported with sirolimus and everolimus. mTOR activation is important for angiogenesis, therefore, it is proposed that sirolimus causes impaired wound healing by inhibiting angiogenesis.59 The mechanism of sirolimus causing impaired wound healing also involves the inhibition of intraepithelial T cells to proliferate and produce normal levels of growth factors. In a mouse model, normal wound closure could be restored by addition of the skin gammadelta T cell-produced factor, insulin-like growth factor-I.60
A prospective randomised trial comparing sirolimus-based immunosuppression versus tacrolimus-based immunosuppression found a significant increase in wound complications in the sirolimus group.61 Tacrolimus is a calcineurin inhibitor and an alternative to sirolimus. Sirolimus also has additional adverse effects including hyperlipidaemia and leukopenia. Dean et al61 adjusted for these in their prospective randomised trial by excluding patients with dyslipidaemia and leukopenia prior to randomisation. However, they were unable to control for pre-existing diabetes mellitus as the sirolimus group had significantly more diabetes compared to the tacrolimus group. In the sirolimus group, 47% (30 of 64) developed wound complications (p<0.0001) compared to 8% of the tacrolimus group (5 of 59). Most wound complications were superficial wound infections, peri-graft fluid collections, and incisional herniae. The sirolimus group had more surgeries and readmissions for wound-related conditions.61
Everolimus is another mTOR inhibitor which reduces wound strength when given at time of surgery. This effect is prevented by delaying administration to 2–4 days after colon surgery in rats.62
Corticosteroids
Corticosteroids are regularly used for their immunosuppressant and anti-inflammatory properties. They include prednisolone, prednisone and dexamethasone and are indicated for the treatment of auto-immune and inflammatory conditions including rheumatoid arthritis, ulcerative colitis, Crohn’s disease, acute asthma and COPD exacerbations and acute gout. Corticosteroids impair wound healing and repair by inhibition of gene expression, anti-inflammatory actions and suppression of multiple cellular wound responses.63 Corticosteroids delay fibroblast proliferation, collagen synthesis, fibroplasia, vascular proliferation and epithelisation.63,64 Their immunosuppressive affects increase wound infection risk which also impedes healing. Corticosteroids used in the inflammatory phase of healing impair leucocyte migration including macrophages into wounds leading to diminished neovascularisation and fibroplasia. Dexamethasone decreases cytokine expression including TNF, PDGF, and IL–1 in damaged wound tissue.64
Colchicine
Colchicine is indicated in acute gout attacks, gout prophylaxis and familial mediterranean fever (FMF). New acute gout guidelines endorse low dose treatment with a reduced adverse risk profile to be as effective as previous high dose treatment; with 1mg taken at the first sign of attack, followed by 500mcg, one hour later.65 Daily prophylactic dosing is once or twice daily and similarly in FMF with daily life-long dosing. Colchicine reduces inflammation, following raised urate crystal levels which commonly deposit in joints and surrounding tissue, providing pain relief.66 Colchicine, an alkaloid, is extracted from plants in the Genus Colchicum.67 Colchicine binds to tubulins with high affinity blocking microtubule assembly and polymerisation.67 Microtubules form complexes used in mitosis, vesicular trafficking and cell motility.68 Successful wound healing requires cell migration into a wound.68 Impaired microtubule assembly and polymerisation impairs cytokine and chemokine secretion, inhibits optimal cell shape maintenance, impairs cell migration and blocks mitotic cell division.67 Colchicine also acts on the immune system inhibiting neutrophil chemotaxis and phagocytosis and adhesion in inflamed tissue.69 Decreased granulocyte migration, decreased fibroblast activity, decreased blood supply due to vasoconstriction and increased collagenase synthesis resulting in a decreased wound breaking strength are all associated with colchicine.53
Antibiotics
Antibiotics treat bacterial infection supporting wound healing in infected wounds. Antibiotics do not heal wounds explicitly as not all wounds are infected, and their indiscriminate use can be harmful.70,71 Correctly identifying infection is key. All open wounds are contaminated with micro-organisms and colonisation occurs when bacteria are replicating without tissue invasion, cellular injury, or wound breakdown.70 Colonised wounds do not require antibiotics.70 Antibiotics treat infection, however they can reduce the tensile strength of wounds, and can affect collagen cross-linking.72
Surprisingly, wound healing is supported in ‘colonised’ wounds by bacteria stimulating neutrophil chemotaxis.71 Proteolytic enzymes from bacteria including hyaluronidases support autolytic debridement and protease release from neutrophils.71
Macrolide antibiotics include erythromycin, clarithromycin and roxithromycin and have diverse biological actions including extensive inflammation modulation and immunomodulatory actions.73 Their use outside their antimicrobial activity in treatment of inflammatory conditions is evolving. Macrolides accumulate within cells and act on receptors that modulate immune cell activities.73 Erythromycin decreases proinflammatory cytokine production, including IL-8 which is a potent neutrophil chemoattractant.73
Tetracycline antibiotics include doxycycline, tetracycline and minocycline. Tetracyclines reduce inflammation by the inhibition of leucocyte chemotaxis and by decreasing pro-inflammatory mediators including TNF and IL-1.74 Tetracyclines inhibit matrix metalloproteinases (MMP) which support wound matrix modification, cell migration and tissue remodelling.75
An exception is doxycycline which has demonstrated positive impacts in the chronic wound environment. Doxycycline inhibits MMP-mediated degradation of a host defense protein (a-1 antitrypsin) which inhibits leucocyte elastase. This reduction in leucocyte elastase indirectly prevents the degradation of connective tissue.76 Nitric oxide (NO) a pro-inflammatory free radical is cytotoxic to the micro-wound environment and overexpressed in chronic wounds. Doxycycline reduces cytokine-induced NO synthesis by inhibition of NO synthase expression reducing excessive tissue breakdown and chronic inflammation.76
Gentamicin is an aminoglycoside which acts to inhibit protein synthesis causing cell membrane damage. It also delays re-epithelialisation in the maturation phase.77
In most wounds topical antibiotic use is not recommended.72 Current 2022 International Wound Infection Institute78 guidelines state, as topical preparations are typically low dose they support resistance and their use should only be employed in critically infected wounds under specialist clinicians.
Dipeptidyl peptidase-4 inhibitors
Dipeptidyl peptidase-4 inhibitors (DPP-4 inhibitors) used in the treatment of type 2 diabetes mellitus have been associated with cutaneous adverse effects. DPP-4 inhibition increases GLP-1 and GIP concentration increasing insulin secretion and inhibits glucagon release to maintain euglycemia.79 DPP-4 inhibitors include alogliptin, linagliptin, sitagliptin, sitagliptin and vildagliptin. DPP-4 is involved in wound healing and immune pathways.80 DPP-4 inhibitors are implicated in the development of cutaneous disease, including Bullous Pemphigoid (BP), mucous membrane pemphigoid and Stevens-Johnson syndrome.81 The average time to BP occurrence is 8-27 months with interpatient variability.81 Rates of BP are higher with vildagliptin, followed by linagliptin.79,81,82 DPP-4 is expressed on immune cell surfaces, including T cells, B cells, macrophages and natural killer cells, endorsing varied immunomodulating affects.82,83 DPP-4 activation on T cells promotes activation and migration, inflammation and autoactivation.80 DPP-4 interacts with other signal transduction pathways (CD3) as a co-stimulator of T cells with activation promoting T cell response to foreign antigens, cytokine secretion, cell proliferation and cellular mobility.84 Fibroblast activation by DPP-4 increases the expression of profibrotic gene expression. Consequently, DPP-4 inhibition inhibits T cell proliferation and helper cell presentation, inhibits keratinocyte DNA synthesis and migration, and suppresses fibroblast survival and proliferation.81
Discussion
Medications are used to improve patient health outcomes by treating disease and symptoms, and sometimes to prolong life in life-threatening conditions. Unfortunately, adverse effects of many medications involve impaired wound healing which can be complicated by necrosis or infection requiring surgery or amputation. Taking a holistic approach to patient management by looking at the patient global wellbeing allows prescribers to treat the condition at hand while remaining cognisant of other current issues which may potentially be negatively affected by the treatment prescribed.
Prior to the prescribing of medication and formulation of a wound management plan, it is essential that a full and complete medication history is taken. Knowledge about medications with potential to cause wounds or delay wound healing aids in reducing these adverse effects from occurring. Medications with long half-lives may still exert their effects when patients are no longer taking them. Identifying previously administered medications can help to mitigate potential negative consequences. For example, perioperative wound complication risk can be reduced by delaying surgery after administration of mTOR inhibitors or VEGF inhibitors or withholding NSAIDs perioperatively. Regular reviews of medications ensures medications with potential negative wound healing impacts are not continued where no longer indicated. Medication dosage and treatment duration also affects the risk of impaired wound healing. Treating an acute gout flare with colchicine for a short duration will have a smaller impact compared to chronic daily dosing for gout prevention.
A pharmacist is the ideal professional to consult prior to prescribing a new medication, especially for patients with wounds. Hospital pharmacies have a dedicated drug information service which can be utilised to support patient-centred prescribing.
Determining the risks and benefits associated with medication use is critical. In some cases, medication use is essential and life-saving and potential associated negative impacts may be accepted, such as in cancer or anticoagulation therapy. Where there are no alternative options available, the choice to use a medication that may impede wound healing may be accepted, but wound prevention and management must be prioritised.
Conflict of interest
The authors declare no conflicts of interest.
Ethics statement
An ethics statement is not applicable.
Funding
The authors received no funding for this study.
Author contribution
All three authors, Giselle Bennett, Julie Abbott, and Geoff Sussman, contributed to literature search, writing and editing.
Author(s)
Giselle Bennett1, Julie Abbott2, Geoff Sussman*3
1Geriatric Registrar Austin Health, Melbourne, Australia
2Pharmacist Quality Pharmacy Group, Melbourne, Australia
3Monash University, Melbourne, Australia
*Corresponding author email Geoff.Sussman@monash.edu
References
- Słonimska P, Sachadyn P, Zieliński J, Skrzypski M, Pikuła M. Chemotherapy-mediated complications of wound healing: An understudied side effect. Adv Wound Care. 2024. https://doi.org/10.1089/wound.2023.0097
- Franz, MG, Steed, DL, Robson, MC. Optimizing healing of the acute wound by minimizing complications. Curr Probl Surg. 2007;44(11):691–763. https://doi.org/10.1067/j.cpsurg.2007.07.001
- Deptuła M, Zieliński J, Wardowska A, Pikuła M. Wound healing complications in oncological patients: perspectives for cellular therapy. Postepy dermatol alergol. 2019;36(2):139-146. https://doi.org/10.5114/ada.2018.72585
- Gao Q, Zhou G, Lin SA-O, Paus R, Yue ZA-O. How chemotherapy and radiotherapy damage the tissue: Comparative biology lessons from feather and hair models. Exp Dermatol. 2019:28(4):413–418. https://doi.org/10.1111/exd.13846
- Regan PO. The impact of cancer and its treatment on wound healing. WOUNDS UK. 2007;3(2):87.
- Bensadoun RJ, Humbert P, Krutman J, Luger T, Triller R, Rougier A, et al. Daily baseline skin care in the prevention, treatment, and supportive care of skin toxicity in oncology patients: recommendations from a multinational expert panel. Cancer Manag Rese 2013; 5:401–408.
- Borenfreund E, Babich H, Martin-Alguacil N. Rapid chemosensitivity assay with human normal and tumor cells in vitro. In vitro cellular & developmental biology. 1990 Nov;26:1030-4. https://doi.org/10.1007/BF02624436
- Castaño E, Rodríguez-Peralto JL, López-Ríos F, Gómez C, Zimmermann M, Iglesias Díez L. Keratinocyte dysplasia: an usual finding after transplantation or chemotherapy. Journal of cutaneous pathology. 2002 Nov;29(10):579-84. https://doi.org/10.1034/j.1600-0560.2002.291002.x
- Buttiglieri S, Ruella M, Risso A, Spatola T, Silengo L, Avvedimento EV, et al. The aging effect of chemotherapy on cultured human mesenchymal stem cells. Exp Hematol. 2011;39(12):1171–1181. https://doi.org/10.1016/j.exphem.2011.08.009
- Kim JA-O, Ohn JA-O, Yoon JS, Kang BM, Park M, Kim S, et al. Priming mobilization of hair follicle stem cells triggers permanent loss of regeneration after alkylating chemotherapy. Nat Commun. 2019;10,3694. https://doi.org/10.1038/s41467-019-11665-0
- Hahn JC, Shafritz AB. Chemotherapy extravasation injuries. J Hand Surg Am. 2012;37(2):360-362. doi: 10.1016/j.jhsa.2011.10.028.
- Schulmeister L. Extravasation management: clinical update. Semin Oncol Nurs. 2011; 27(1):82-90. doi: 10.1016/j.soncn.2010.11.010
- Sirieix M-E, Debure C, Baudot N, Dubertret L, Roux ME, Morel P, et al. Leg ulcers and hydroxyurea: forty-one cases. Arch Dermatol. 1999;135(7):818-820.
- Best PJ, Daoud MS, Pittelkow MR, Petitt RM. Hydroxyurea-induced leg ulceration in 14 patients. Ann Intern Med. 1998; 128(1):29–32.
- Antonioli E, Guglielmelli P, Pieri L, Finazzi M, Rumi E, Martinelli V, et al. Hydroxyurea-related toxicity in 3,411 patients with Ph’-negative MPN. Am Journal Hematol. 2012; 87(5):552–554. https://doi.org/10.1002/ajh.23160
- Er O, Inanc M, Dikilitas M, Ozkan M, Karaca H, Saraymen R, et al. VEGF, EGF, HGF, and PDGF levels during chemotherapy in patients with metastatic colorectal cancer. J Clinical Oncol. 2009;27(15,sup): 4057.
- Bates DO, Jones ROP. The role of vascular endothelial growth factor in wound healing. Int J Low Extrem Wounds. 2003;2(2):107-120.
- Zhang H, Huang Z, Zou X, Liu T. Bevacizumab and wound-healing complications: a systematic review and meta-analysis of randomized controlled trials. Oncotarget. 2016;7(50):82473-82481. doi: 10.18632/oncotarget.12666.
- Scappaticci FA, Fehrenbacher L, Cartwright T, Hainsworth JD, Heim W, Berlin J, et al. Surgical wound healing complications in metastatic colorectal cancer patients treated with bevacizumab. J Surg Oncol (Tallinn). 2005;91(3):173-180.
- Sharma K, Marcus JR. Bevacizumab and wound-healing complications: mechanisms of action, clinical evidence, and management recommendations for the plastic surgeon. Ann Plast Surg. 2013;71(4):434-440.
- Taira K, Nadatani Y, Hirano S, Maeda K, Fujiwara Y. Large skin ulcer and delayed wound healing around a colostomy in a patient with metastatic colorectal cancer receiving Vascular Endothelial Growth Factor Receptor-2 Inhibitor Therapy. Case Rep Oncol. 2019;12(2):370-375.
- Sindt JE, Fitzgerald LA, Kuznicki J, Prelewicz S, Odell DW, Brogan SE. Antiplatelet and wound healing implications of immunotherapy and targeted cancer therapies in the perioperative period. Anesthesiology. 2023;139(4):511-522.
- Matsuo M, Hashimoto K, Jiromaru R, Nakagawa T. Delayed pharyngocutaneous fistula caused by molecular targeted therapy: a case report. J Med Case Reports. 2022;16:383. https://doi.org/10.1186/s13256-022-03621-2
- Roger A, Sigal ML, Bagan P, Sin C, Bilan P, Dakhil B, et al. Ulcères des membres inférieurs développés sous inhibiteurs de tyrosine kinase (sunitinib, nilotinib) [Leg ulcers occurring under tyrosine kinase inhibitor therapy (sunitinib, nilotinib)]. Annales de dermatologie et de venereologie. 2017;144(1):49–54.
- Carvalho FLF, Zheng C, Witmer K, O’Neill J, Lynch JH, Kowalczyk KJ. Complications associated with perioperative use of tyrosine kinase inhibitor in cytoreductive nephrectomy. Sci Rep. 2019;9(1):15272.
- Abd-El-Aleem SA, Ferguson MWJ, Appleton I, Bhowmick A, McCollum CN, Ireland GW. Expression of cyclooxygenase isoforms in normal human skin and chronic venous ulcers. The J Pathology. 2001;195(5):616-623.
- Elder CL, Dahners LE, Weinhold PS. A cyclooxygenase-2 inhibitor impairs ligament healing in the rat. Am J Sports Med. 2001 Nov-Dec;29(6):801-805. doi: 10.1177/03635465010290062101
- Tsai WC, Hsu CC, Chen CP, Chen MJ, Lin MS, Pang JH. Ibuprofen inhibition of tendon cell migration and down-regulation of paxillin expression. J Orthop Res. 2006 Mar;24(3):551-558. doi: 10.1002/jor.20069
- Tsai WC, Hsu CC, Chou SW, Chung CY, Chen J, Pang JH. Effects of celecoxib on migration, proliferation and collagen expression of tendon cells. Connect Tissue Res. 2007;48(1):46-51. doi: 10.1080/03008200601071295.
- Dimmen S, Nordsletten L, Engebretsen L, Steen H, Madsen JE. The effect of parecoxib and indometacin on tendon-to-bone healing in a bone tunnel: an experimental study in rats. J Bone Joint Surg Br. 2009 Feb;91(2):259-263.
- Connizzo BK, Yannascoli SM, Tucker JJ, Caro AC, Riggin CN, Mauck RL, Soslowsky LJ, Steinberg DR, Bernstein J. The detrimental effects of systemic Ibuprofen delivery on tendon healing are time-dependent. Clinical Orthopaedics and Related Research®. 2014 Aug;472:2433-9. doi: 10.1007/s11999-013-3258-2
- Mallick E, Scutt N, Scutt A, Rolf C. Passage and concentration-dependent effects of Indomethacin on tendon derived cells. Journal of Orthopaedic Surgery and Research. J Orthop Surg Res. 2009 Apr 2;4:9. doi: 10.1186/1749-799X-4-9
- Ad-El DD, Weinberg A, Kogan L, Arieli D, Neuman A, Meirovitz A, et al. Warfarin skin necrosis: local and systemic factors. Br J Plast Surg. 2000;53(7):624-626.
- Eby CS. Warfarin-induced skin necrosis. Hematol Oncol Clin North Am. 1993;7(6):1291-1300.
- Kipen CS. Gangrene of the breast: A complication of anticoagulant therapy: Report of two cases. N Eng J Med. 1961;265(13):638-640.
- Verhagen H. Local haemorrhage and necrosis of the skin and underlying tissues, during anti-coagulant therapy with dicumarol or dicumacyl. Acta Med Scand. 1954;148(6):453‑468.
- Nalbandian RM, Mader IJ, Barrett JL, Pearce JF, Rupp EC. Petechiae, ecchymoses, and necrosis of skin induced by coumarin congeners: rare, occasionally lethal complication of anticoagulant therapy. JAMA. 1965;192(7):603-608.
- Beitz JM. Coumadin-induced skin necrosis. Wounds. 2002;14(6):217-20.
- Berkompas DC. Coumadin skin necrosis in a patient with a free protein S deficiency: case report and literature review. Indiana Med. 1991;84(11):788-91.
- Brooks LW, Blais FX. Coumarin-induced skin necrosis. J Am Osteopath Assoc. 1991;91(6):601-5.
- DeFranzo AJ, Marasco P, Argenta LC. Warfarin-induced necrosis of the skin. Ann Plast Surg. 1995;34(2):203-8.
- Chan YC, Valenti D, Mansfield AO, Stansby G. Warfarin induced skin necrosis. Br J Surg. 2000;87(3):266-272.
- Gelwix TJ, Beeson MS. Warfarin-induced skin necrosis. Am J Emerg Med. 1998;16(5):541-543.
- Rose VL, Kwaan HC, Williamson K, Hoppensteadt D, Walenga J, Fareed J. Protein C antigen deficiency and warfarin necrosis. Am J Clin Pathol. 1986;86(5):653-655.
- Cole MS, Minifee PK, Wolma FJ. Coumarin necrosis—a review of the literature. Surgery. 1988;103(3):271-7.
- Dissemond J. Skin Necrosis and Ulcers Induced by Medications. In: Téot, L., Meaume, S., Akita, S., Ennis, W.J., del Marmol, V. (editors) Skin Necrosis. Vienna: Springer; 2015. p. 95-100 https://doi.org/10.1007/978-3-7091-1241-0_14
- Vu TT, Gooderham M. Adverse drug reactions and cutaneous manifestations associated with anticoagulation. J Cutan Med Surg. 2017;21(6):540-550.
- Cakmak MA, Sahin S, Cinar N, Karsidag S. Adverse skin reaction caused by dabigatran. 2014.
- Potolidis E, Mandros C, Kotsa K, Mitsiou E, Potolidis D, Fanourgiakis P. Dabigatran associated leukocytoclastic vasculitis. Case Rep Med. 2015. doi: 10.1155/2015/616109
- Tsoumpris A, Tzimas T, Gkabrelas K, Akritidis N. Iron complex, dabigatran and toxic epidermal necrolysis syndrome: a case report. J Clin Pharm Ther. 2013;38(2):177-178.
- Barrett P, Vuppalanchi R, Masuoka H, Chalasani N. Severe drug-induced skin and liver injury from rivaroxaban. Dig Dis Sci. 2015;60:1856-1858.
- Pansuriya T, Nguyen T, Sadat MA, Raza SA, Sarva ST. A 78-year-old man with a pulmonary embolism who developed skin necrosis 7 days after treatment with the direct oral anticoagulant factor Xa inhibitor apixaban. Am J Case Rep. 2021;22:e929002-1.
- Karukonda SR, Flynn TC, Boh EE, McBurney EI, Russo GG, Millikan LE. The effects of drugs on wound healing. Part II. Specific classes of drugs and their effect on healing wounds. Int J Dermatol. 2000;39(5):321-333.
- Delyon J, Ortonne N, Benayoun E, Moroch J, Wolkenstein P, Sbidian E, et al. Low-dose methotrexate-induced skin toxicity: Keratinocyte dystrophy as a histologic marker. J Am Acad Dermatol. 2015 Sep;73(3):484-490.
- Bootun R. Effects of immunosuppressive therapy on wound healing. Int Wound J. 2013;10(1):98-104.
- McCoy CM. Leflunomide-Associated Skin Ulceration. Ann Pharmacother. 2002;36(6):1009-1011.
- Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;6:15.
- Rozman B. Clinical pharmacokinetics of leflunomide. Clin pharmacokinet. 2002;41(6):421-30.
- Humar ROK, Kiefer FN, Berns H, Resink TJ, Battegay EJ. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. The FASEB Journal. 2002;16(8):771-780.
- Mills RE, Taylor KR, Podshivalova K, McKay DB, Jameson JM. Defects in skin gamma delta T cell function contribute to delayed wound repair in rapamycin-treated mice. J Immunol. 2008 Sep 15;181(6):3974-3983. doi: 10.4049/jimmunol.181.6.3974
- Dean PG, Lund WJ, Larson TS, Prieto M, Nyberg SL, Ishitani MB, et al. Wound-healing complications after kidney transplantation: a prospective, randomized comparison of Sirolimus and Tacrolimus1. Transplantation. 2004;77(10):1555-1561.
- Willems MC, Hendriks T, de Man BM, Lomme RM, van der Vliet JA. Everolimus-induced loss of wound strength can be prevented by a short postoperative delay in its administration. Wound Repair Regen. 2011 Nov;19(6):680-686. doi: 10.1111/j.1524-475X.2011.00730.x.
- Guo S, Dipietro LA. Factors affecting wound healing. J Dent Res. 2010 Mar;89(3):219-229. doi: 10.1177/0022034509359125
- Weinberg E, Tagger-Green N, Lusthaus M, Vered M, Mijiritsky E, Chaushu L, et al. The Impact of Corticosteroid Administration at Different Time Points on Mucosal Wound Healing in Rats: An Experimental Pilot In Vivo Study. Biology. 2022;11(9):1309.
- Finch A, Kubler P. The management of gout. Aust Prescr. 2016 Aug;39(4):119-122. doi: 10.18773/austprescr.2016.047
- Coburn BW, Mikuls TR. Treatment Options for Acute Gout. Fed Pract. 2016 Jan;33(1):35-40.
- Leung YY, Yao Hui LL, Kraus VB. Colchicine: Update on mechanisms of action and therapeutic uses. Semin Arthritis Rheum. 2015 Dec;45(3):341-350. doi: 10.1016/j.semarthrit.2015.06.013
- Charafeddine RA, Nosanchuk JD, Sharp DJ. Targeting Microtubules for Wound Repair. Adv Wound Care. 2016 Oct 1;5(10):444-454. doi: 10.1089/wound.2015.0658.
- Cronstein BN, Molad Y, Reibman J, Balakhane E, Levin RI, Weissmann G. Colchicine alters the quantitative and qualitative display of selectins on endothelial cells and neutrophils. J Clin Invest. 1995 Aug;96(2):994-1002. doi: 10.1172/JCI118147
- Caldwell MD. Bacteria and Antibiotics in Wound Healing. Surg Clin North Am. 2020;100(4):757-776.
- de Souza JM, Vieira ÉC, Cortez TM, Mondelli AL, Miot HA, Abbade LPF. Clinical and microbiologic evaluation of chronic leg ulcers: a cross-sectional study. Adv Skin Wound Care. 2014;27(5):222-227.
- Diehr S, Hamp A, Jamieson B, Mendoza M. Clinical inquiries. Do topical antibiotics improve wound healing? J Fam Pract. 2007;56(2):140-144.
- Kanoh S, Rubin BK. Mechanisms of Action and Clinical Application of Macrolides as Immunomodulatory Medications. Clin Microbiol Rev. 2010;23(3):590-615.
- Pasquale TR, Tan JS. Nonantimicrobial effects of antibacterial agents. Clin Infect Dis. 2005;40(1):127-135.
- Caley MP, Martins VLC, O’Toole EA. Metalloproteinases and Wound Healing. Adv in Wound Care (New Rochelle). 2015;4(4):225-234.
- Wilcox JR, Covington DS, Paez N. Doxycycline as a modulator of inflammation in chronic wounds. Wounds. 2012;24(12):339-349.
- Berry JAD, Miulli DE, Lam B, Elia C, Minasian J, Podkovik S, et al. The neurosurgical wound and factors that can affect cosmetic, functional, and neurological outcomes. Int Wound J. 2019;16(1):71-78.
- Swanson T, Ousey K, Haesler E, Bjarnsholt T, Carville K, Idensohn P, et al. IWII Wound Infection in Clinical Practice consensus document: 2022 update. J Wound Care. 2022 Dec 1;31(Sup12):S10-S21. doi: 10.12968/jowc.2022.31.Sup12.S10
- Gallwitz B. Clinical use of DPP-4 inhibitors. Front Endocrinol (Lausanne). 2019;10:389.
- Zhang K-W, Liu S-Y, Jia Y, Zou M-L, Teng Y-Y, Chen Z-H, et al. Insight into the role of DPP-4 in fibrotic wound healing. Biomed Pharmacother. 2022 Jul;151:113143. doi: 10.1016/j.biopha.2022.113143
- Patel PM, Jones VA, Kridin K, Amber KT. The role of Dipeptidyl Peptidase-4 in cutaneous disease. Exp dermatol. 2021;30(3):304-318.
- Kawaguchi Y, Shimauchi R, Nishibori N, Kawashima K, Oshitani S, Fujiya A, et al. Dipeptidyl peptidase-4 inhibitors-associated bullous pemphigoid: A retrospective study of 168 pemphigoid and 9,304 diabetes mellitus patients. J Diabetes Investig. 2019;10(2):392-398.
- Huang J, Liu X, Wei Y, Li X, Gao S, Dong L, et al. Emerging Role of Dipeptidyl Peptidase-4 in Autoimmune Disease. Front Immunol. 2022 Mar 4;13:830863. doi: 10.3389/fimmu.2022.830863
- Lee J-G, Kang DG, Yu JR, Kim Y, Kim J, Koh G, et al. Changes in Adenosine Deaminase Activity in Patients with Type 2 Diabetes Mellitus and Effect of DPP-4 Inhibitor Treatment on ADA Activity. Diabetes Metab J. 2011;35(2):149-158.

