

Volume 30 Number 1
Pyoderma gangrenosum: a review of the clinical, mechanistic and therapeutic landscape
Akshay Flora and John W Frew
Keywords Pyoderma gangrenosum, Inflammation, Th17, neutrophils
For referencing Flora A and Frew JW. Pyoderma gangrenosum: a review of the clinical, mechanistic and therapeutic landscape. Wound Practice and Research 2022; 30(1):16-23.
DOI
https://doi.org/10.33235/wpr.30.1.16-23
Submitted 21 September 2021
Accepted 22 October 2021
Abstract
Pyoderma gangrenosum (PG) is a neutrophilic dermatosis that is uncommon and can sometimes be associated with systemic diseases. The pathophysiology underlying this condition is poorly understood, although recent advances suggest that local cutaneous abnormalities and functionally abnormal neutrophils may trigger ongoing innate and adaptive immune system activity. PG remains a difficult condition to diagnose, mainly because it was previously seen as a diagnosis of exclusion, although newer diagnostic criteria have been proposed in order to overcome this. Furthermore, many patients do not respond to conventional therapy for PG once diagnosed, and experience persistence or worsening of their condition over time. The advent of immune targeted therapies, however, may represent a new treatment option for these patients. This review focuses on the clinical features and diagnosis of PG, as well as providing an update in our understanding of the pathophysiology and treatment options available for this debilitating condition.
Introduction
Pyoderma gangrenosum (PG) is a painful and ulcerative condition that is classified as a neutrophilic dermatosis. It is an uncommon disease, with a worldwide incidence of approximately 3–10 cases per million population per year1. Although it may occur at any age, it is mostly seen in those between 20–50 years of age, with females being affected slightly more than males.
The condition was first described in 1908 by Louis Brocq, and named as pyoderma gangrenosum in 1930 by Brunsting et al.2. The term ‘pyoderma’ refers to a purulent infection of the skin, whilst ‘gangrenosum’ refers to the extensive necrosis seen in these ulcers. It was later found, however, that these ulcers are primarily aseptic in nature, and hence the condition being referred to as ‘pyoderma’ is a misnomer.
The pathophysiology underlying PG is yet to be known, although it may occur secondary to other inflammatory diseases. PG was previously thought to arise from a functional disorder of neutrophils, although recent advances suggest there is also adaptive and innate immune system dysregulation, as well as local cutaneous abnormalities.
Due to the lack of understanding behind how PG develops, there is yet to be a highly effective treatment which targets biological pathways. Current management involves optimal wound care and topical or systemic steroids or steroid sparing agents. Certain biological agents, including IL‑23 and IL‑17 antagonists, as well as JAK-STAT inhibitors, however, may hold promise in the rapid treatment of this condition.
Clinical features
PG most commonly occurs on the lower extremities, although other areas, including the trunk, abdomen, scalp and face, may be affected. It may also affect extra-cutaneous locations, including the lungs, eyes and mouth (termed pyostomatitis vegetans)3.
Up to 25% of patients affected with PG will experience ‘pathergy’ or the Koebner phenomenon4. This occurs when localised trauma causes worsening of existing PG or the formation of new PG lesions. The mechanism underlying pathergy remains poorly understood, although a similar process has also been described in Behcet’s disease5.
Subtypes
Ulcerative PG (classic form)
This type of PG initially starts as either a deep and tender nodule or a superficial pustule which undergoes necrosis and ulceration, usually over the course of a few days6. Following this, the ulcer may follow one of two clinical courses. It may either rapidly expand to involve previously unaffected surrounding tissue, along with severe pain and systemic symptoms6. Alternatively, it may gradually spread over the course of months, with some areas undergoing spontaneous resolution, and other areas undergoing further growth6.
During ulcer expansion, the border is often elevated with an undermined edge, and has a dull red or violaceous appearance (Figure 1A). An erythematous ‘halo’ may be seen surrounding this border, signalling active inflammation in adjacent areas of skin. The base of the ulcer is usually necrotic and has purulent exudate or small abscesses6.
Pustular PG
Pustular PG is characterised by crops of painful pustules which, unlike ulcerative PG, do not undergo ulcer formation7. The pustules are usually accompanied with fevers, arthralgias and joint effusions7. This form of PG also appears to be associated with ulcerative colitis (UC)7. Pustular PG severity does not appear to be related to UC severity however, and treatment of the UC does not necessarily improve the PG8.
Bullous PG
Also known as atypical PG, this form is characterised by rapid superficial skin necrosis with overlying blister formation9. A grey hue is often noted in surrounding tissue, and the lesions are painful; rupture or removal of the bulla may reveal a superficial ulcer (Figure 1B). The bullae are typically located on the arms and face, as opposed to the lower extremities9. It has been reported in patients with haematological disorders9.
Sweet’s syndrome (acute febrile neutrophilic dermatosis) is believed to be on the same spectrum as bullous PG as it too can present with superficial erosions9. However, Sweet’s syndrome lesions present as plaques or nodules and, unlike PG, lack a violaceous undermined border10.
Vegetative PG
Vegetative PG is characterised by a verrucous appearance, with shallow ulcers that lack an undermining border and which usually have no purulent exudate within the base (Figure 1C). They are usually located on the head and neck region and are usually indolent in nature9. There is, however, a rare and more aggressive form of vegetative PG known as malignant PG. Whilst these ulcers are also located on the head and neck, they rapidly expand and may erode underlying structures including the parotid gland11.
Peristomal PG
This type of PG occurs in patients who have an ileostomy or colostomy for underlying inflammatory bowel disease (IBD). It is localised to surrounding areas of the stoma (Figure 1D) and is believed to occur due to ongoing irritation from faecal material passing through the stoma, or from adhesives that are used to attach the stoma bag to the stoma itself.
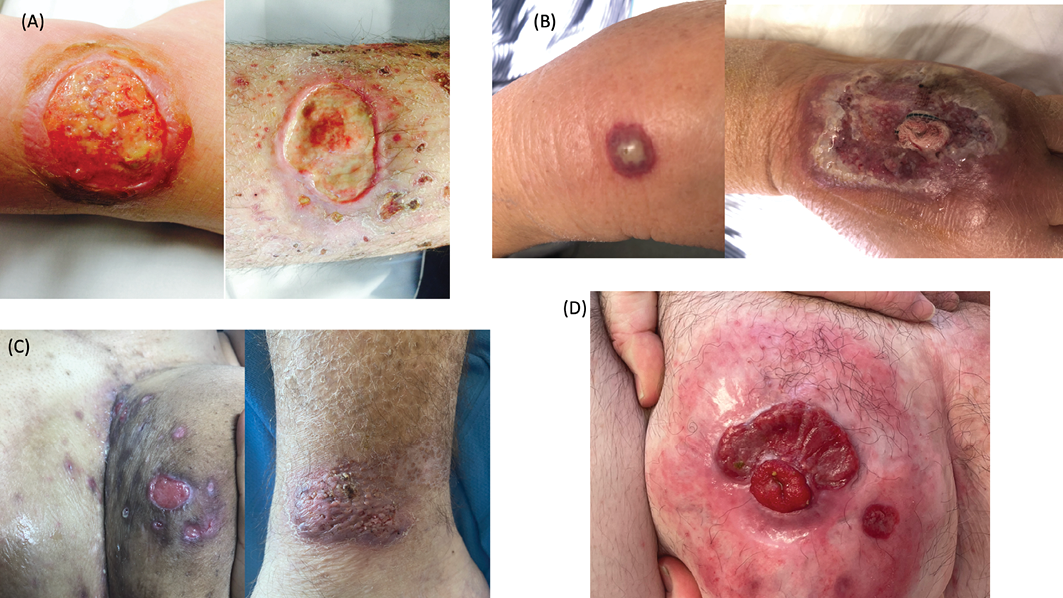
Figure 1. Types of PG
A=ulcerative (classic) PG on the lower leg
B=bullous PG on the dorsal hand, rapidly progressing to an undermined violaceous-bordered painful ulcer
C=painful PG lesions on the buttock of an individual with hidradenitis suppurativa and IBD, and a verrucous plaque of vegetative PG on the lateral ankle
D=peristomal PG in the setting of Crohn’s disease
Systemic associations
Approximately 25–50% of patients with PG will have an underlying inflammatory systemic disease. The systemic disease may be subclinical, and may occur before or after the onset of PG. It is therefore important to appropriately investigate and monitor patients with PG for these conditions.
IBD is the most commonly associated disease, with one study finding it present in 41% of PG patients12. Although the exact reason behind this association remains unknown, common gene mutations in PG and IBD have been identified with respect to antigen presentation and cytokine signalling, including TIMP3 and IL8RA13. Furthermore, alterations in the Wnt signalling pathway, which is known to occur within the intestinal tract of IBD patients14, may also be occurring within PG ulcers. The Wnt pathway is crucial in maintaining epidermal stem cells for reepithelisation during tissue damage14. Wnt signalling has been found to be suppressed in the wounds of diabetic patients15,16, and a similar process may be occurring in PG, although further studies are required. Interestingly, PG activity does not necessarily correlate with the activity of underlying IBD, and suppression of IBD will not necessarily lead to a reduction in PG activity or ulceration.
Rheumatoid arthritis also affects approximately 8.5% of patients with PG12. Other inflammatory arthritis that has been associated with PG include psoriatic arthritis, ankylosing spondylitis and synovitis, acne, pustulosis, hyperostosis and osteitis (SAPHO) syndrome, albeit at a much lower rate.
A vast range of haematological malignancies have also been associated with PG, including monoclonal gammopathy, myeloma, leukaemia, lymphoma and myelodysplasia12. PG as a paraneoplastic phenomenon has also been described in the literature, including in cancer and neuroendocrine tumours17,18.
Associated syndromes
Certain autoinflammatory syndromes are characterised by the presence of PG. The mechanism underlying these syndromes appears to be mediated by the over-expression of IL‑1B19. IL‑1B causes the release of inflammatory cytokines including TNF‑α, IFN‑γ, IL‑8 and Regulated on Activation, Normal T-Cell Expressed and Secreted (RANTES)20. IL‑1B also prevents apoptosis of neutrophils, enabling for ongoing tissue destruction21.
Pyogenic Arthritis, Pyoderma gangrenosum and Acne (PAPA) syndrome is associated with mutations in the PSTPIP1 gene22 which enables overactivation of the inflammasome and subsequent cleavage of pro IL‑1B into IL‑1B23. The syndrome is characterised by a recurring, sterile, monoarticular arthritis along with severe nodulocystic acne and PG23.
Pyoderma gangrenosum, Acne and Suppurative Hidradenitis (PASH) syndrome may be associated with CCTG motif repeats near the PSTPIP1 promoter region19, again enabling for overactivation of the inflammasome. However, there is new evidence to suggest that the cause is polygenic24.
Pyogenic Arthritis, Acne, Pyoderma gangrenosum and Suppurative Hidradenitis (PAPASH) is another rare syndrome that lacks genetic studies, although a p.E227D missense mutation within exon 10 and 11 of the PSTPIP1 gene has been identified within one patient19. The significance of this mutation is unknown, and further studies in patients are required.
Diagnosis and investigations
The diagnosis of PG can often be challenging, with many cases being initially misdiagnosed25. This is because PG shares some overlapping features with other diseases, and there are no diagnostic histological or laboratory investigations. PG has traditionally been a diagnosis of exclusion although, recently, there have been two proposed criteria for PG diagnosis known as the Delphi consensus and the PARACELSUS score.
The Delphi consensus (Table 1) has a sensitivity of 86% and specificity of 90%26. It requires identification of neutrophilic infiltrate within biopsy of the ulcer edge as its sole major criteria26. At least four of the eight minor criteria must also be satisfied, including pathergy, exclusion of infection, a history of IBD or inflammatory arthritis, ulceration of a papule/pustule/vesicle within 4 days of appearance, undermining border/peripheral erythema/ulcer tenderness, presence of multiple ulcers with at least one being on the anterior lower leg, cribriform scarring, or a reduction in ulcer size 1 month after commencing immunosuppressants26.
Table 1. The Delphi consensus for the diagnosis of ulcerative PG26
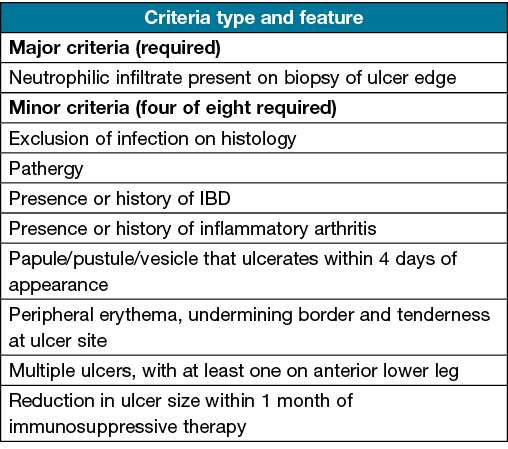
The histological requirement of a neutrophilic infiltrate for PG in the DELPHI criteria has been subject to criticism, as these findings are usually present in the acute phase of PG27. A retrospective study identified only 7% of PG patients had this characteristic finding28. Furthermore, with respect to cribriform scarring being part of the DELPHI minor criteria, one recent study analysing 62 PG scars identified no evidence of any cribriform type scarring, suggesting that it may not actually be associated with PG29.
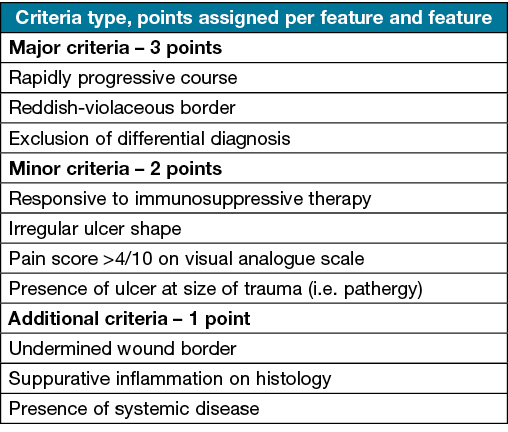
The PARACELSUS scoring system (Table 2) requires at least 10 points for the diagnosis of PG30. There are three major criteria each assigned three points, and include disease progression, exclusion of differential diagnosis and a reddish violaceous wound border30. The minor criteria assigned two points include responsiveness to immunosuppression, irregular ulcer shape, pain greater than 4 on visual analogue scale, or localisation of PG at sites of trauma30. Additional criteria are worth one point and include undermined wound border, systemic disease involvement and presence of suppurative inflammation30. Overall sensitivity and specificity data for this scoring system has not been stated.
Regardless of the criteria used, a biopsy is highly recommended in order to help exclude other conditions with a similar clinical appearance25. Whilst a biopsy does risk pathergy if the lesion is truly PG, it is generally outweighed by the need to reach an accurate diagnosis and commence appropriate treatment25.
Table 2. The PARACELSUS score for the diagnosis of PG30: a score of 10 or above indicates that PG is highly likely
Other relevant investigations
Patients with PG may have leucocytosis and elevated inflammatory markers including C-reactive protein and erythrocyte sedimentation rate6. Swabs for culture should also be taken from PG ulcers, although growth may be indicative of a secondary wound colonisation rather than infection.
Further investigations may also be necessary if an underlying systemic disease is suspected6. Patients who have features suggestive of IBD should be promptly referred to a gastroenterologist for consideration of a colonoscopy and further management. Those with features of inflammatory arthropathy should be referred to a rheumatologist, along with testing for rheumatoid factor and anti-cyclic citrullinated peptide antibodies. If a haematological malignancy is considered, serum protein electrophoresis and serum and urine immunoelectropheresis may also assist in the diagnosis.
Although less common, systemic lupus erythematosus31 and vasculitis32 may also cause PG, in which case anti-nuclear antibodies and anti-neutrophilic cytoplasmic antibodies should be performed.
Differential diagnosis
The differential diagnosis is broad, and includes conditions that may present with ulceration. These include vascular occlusion and venous ulcers, malignancies (including primary cutaneous lymphomas), systemic vasculitis (including granulomatosis with polyangiitis), cutaneous infections, external trauma, drug reactions and other neutrophilic dermatoses.
Pathophysiology
The exact mechanism through which PG arises remains poorly understood33. The rapidly progressive and ulcerative nature of this disease from seemingly ‘normal’ skin makes it difficult to identify early initiating events that may lead to downstream inflammatory cascades33. Histologically, neutrophils are known to predominate within established PG, but it is unknown whether their presence is due to primary neutrophilic abnormalities or is secondary to an already established complex immunological dysfunction34.
Neutrophil activity in PG
During infections, neutrophils normally function to produce extracellular traps, a meshwork consisting of chromatin fibres and degradative enzymes which trap and destroy microbes. Neutrophil extracellular traps (NETs) have been found to be elevated, particularly within the serum of patients with syndromic forms of PG35,36. The neutrophils are functionally abnormal in that there is an enhanced propensity for spontaneous NET formation and reduced ability to degrade NETs35. The ongoing presence of NETs within tissue may prime B cells to produce autoantibodies against certain NET components as seen within other conditions such as hidradenitis suppurativa, and thus cause antibody mediated inflammation37.
A variety of cytokines related to neutrophil activity have also been identified in PG tissue. Dermal fibroblasts, endothelial cells and other local immune cells may be releasing IL‑8 which serves as a chemoattractant for circulating neutrophils20,38. The increased expression of chemokine (C-X-C motif) ligand (CXCL) 1/2/3, CXCL16 and RANTES allows for circulating neutrophils to migrate through the vascular endothelium into PG tissue20,39,40, whilst TNF‑α39 and C5a41 enables sustenance and amplification of neutrophilic activity. Once neutrophils enter the tissue, destruction and ulceration is likely facilitated through the over expression of metalloproteinases including MMP-2 and MMP-8 which disrupts the extracellular matrix20,39,42 and leads to necrosis.
Adaptive immune system activity
The Th17 pathway has been strongly associated with PG activity. This has been identified through the overexpression of IL‑17 and its receptor within PG tissue from multiple translational studies35,38,39, as well as through the rapid and sustained clinical improvement seen with IL‑23 and IL‑17 antagonists within PG case reports43–45. It is possible that Th17 may also be activated through the release of IL‑9 from local Th9 cells (as seen with other inflammatory dermatoses)46, in addition to IL‑23 pathways.
IL‑12 has also been identified to be expressed in PG tissue, likely leading to the stimulation of the Th1 response and subsequent release of IFN‑γ and CXCR3, enabling for leucocyte recruitment and differentiation38,47. Similarly, elevated IL‑4 levels within tissue may be driving Th2 differentiation, causing IL‑5, IL‑13 and CCR3 release47. These cytokines are known to stimulate B cells to produce antibodies, including IgA and IgE, which may contribute to further tissue destruction.
Regulatory T-cell (Treg) activity may also be reduced within PG lesions, with a reduction in the FOXP3/RORyt ratio, TGF‑β/CD4+ ratio and IL‑10/CD4+ ratio being identified within one translational study48. The increased Th1, Th2 and Th17 response coupled with reduced Treg activity indicates that T-cell-mediated inflammation plays a substantial role in PG pathogenesis.
Follicular adnexal structures in PG
PG ulcers generally do not affect areas of the body lacking follicular adnexal structures, including the palms and soles33. Areas of skin that have previously been affected by PG and have undergone fibrosis (along with an absence of follicular adnexal structures), also appear to be resistant to PG re-ulceration33. It is possible that the development of auto-antigens to components of the follicular adnexa may be a key initiating event for the development of PG33. The lack of CD34+ (fibroblast) cells identified in PG scar biopsies33 when compared to active PG ulcers may indicate that fibroblasts play an inflammatory role in active PG ulceration.
Gene expression studies
The recent use of RNA sequencing has enabled for an analysis of gene expression studies in patients with PG when compared to healthy controls. One recent study of eight PG patients with perilesional biopsies identified 5,762 genes that were differentially expressed to a significant extent when compared to healthy control biopsies49. Furthermore, within perilesional PG tissue of these patients, there was a large upregulation of inflammatory cytokine related genes in the dermis and a downregulation of these genes in the epidermis49. Several inflammatory and trafficking pathways have been identified within this study which may play a role in PG pathogenesis, although further studies are required to more comprehensively characterise this49.
Treatment
There is yet to be a universal, standardised treatment approach for PG. Whilst a range of topical and systemic therapeutic options are available, they have all demonstrated variable success across patients.
Stratification of PG into mild, moderate and severe forms may assist in treatment choice. The number and location of PG ulcers, ulcer size, rate of ulcer expansion, as well as extracutaneous PG involvement, should all be considered when determining disease severity. If PG has occurred secondary to a systemic disease, then it is imperative that the systemic disease is adequately treated, as this too may lead to ulcer improvement. A combination of local and systemic treatments, along with regular wound care, has been found to confer the highest likelihood of adequate ulcer healing50.
Topical therapies
Topical treatments include tacrolimus ointment 0.1% or super potent topical steroids, including clobetasol propionate51,52. Tacrolimus is a calcineurin inhibitor and functions by inhibiting the expression or transcription of genes encoding IL‑2, IL‑3, IL‑4, IL‑8, TNF‑α and granulocyte-macrophage colony stimulating factor (GM-CSF)53. These cytokines are predominant in T-cell activation and cytotoxicity54, hence their inhibition enables for a dampening of the inflammatory response seen within PG ulcers. A study comparing these two treatments for peristomal PG found that topical tacrolimus was associated with a higher rate of PG healing55. Intralesional steroid injections with triamcinolone acetonide 6–40mg/L into the active borders of the PG ulcer may also halt ulcer expansion and promote healing56.
Systemic therapies
With regards to systemic therapies, oral prednisolone and cyclosporine are the most commonly used first line treatments. Oral prednisolone is typically commenced at 0.5–1mg/kg whilst cyclosporine is dosed at 3–5mg/kg, with both being found to have approximately the same rate of ulcer healing 6 weeks post-initiation57. Where prednisolone is being used as a first line agent, the dosage may be weaned once appropriate control of PG has been achieved, with consideration of transitioning to a steroid sparing agent including cyclosporine, azathioprine, mycophenolate mofetil or methotrexate. Other anti-inflammatory adjunctive agents, including colchicine, dapsone and tetracyclines, have been used as part of combination systemic therapy for moderate to severe PG. In cases of rapidly progressive PG, the use of intravenous corticosteroids, including pulsed methylprednisolone of intravenous immunoglobulin (IVIG), may be considered58.
Biological therapies
Biological therapies are a suitable option for PG refractory to systemic and topical therapies. Due to the rarity of PG, the use of biological agents has been mostly limited to small observational studies or case reports. Hence, further studies are required to further evaluate their efficacy in treating this condition.
TNF‑α antagonists have been the most widely studied agent for PG as they are also commonly used to treat coexisting IBD. Infliximab, adalimumab and etanercept have all demonstrated efficacy through a reduction in ulcer size and improvement in related symptoms59–61.
IL‑23 inhibitors, including ustekinumab (IL‑12/IL23p40) and guselkumab, as well as IL‑17 inhibitors, including brodalumab, have also demonstrated effectiveness in PG ulcer healing43–45. This is likely due to inhibition of the Th17 axis which has been implicated as a major contributor to PG pathogenesis.
JAK-STAT inhibitors, including tofacitinib, may represent a new and effective treatment option for PG as they are able to downregulate the production of multiple associated cytokines including IL‑23R, IL‑12R, and IL‑10R. One study of three patients with PG associated with Crohn’s disease and inflammatory arthritis found marked ulcer healing and symptoms improvement within 12 weeks of treatment with tofacitinib62.
Wound care
Whilst there are no specific guidelines for optimal wound care in PG, the main goals are to protect the ulcers from experiencing further physical trauma and fostering a microenvironment that enables wound healing. The type of dressings used may be guided by the tissue, infection, moisture balance and edge advancement (TIME) approach for chronic wounds63 (Table 3).
Table 3. Treatment options available for different types of PG
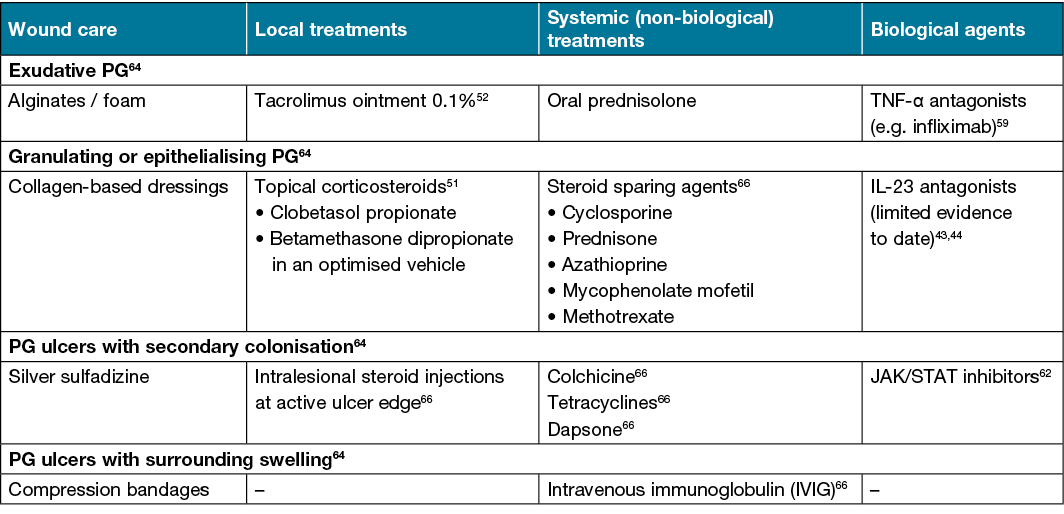
Active ulcers will usually produce large amounts of exudate due to high neutrophil activity, which leaves the surrounding normal skin at risk of maceration and infection. Alginate dressings enable for high amounts of fluid absorption whilst still providing adequate moisture to the wound64. Foam dressings are useful for affected areas of skin that may be subject to physical trauma, although their absorptive ability is lower than alginates64. PG wounds that are epithelising or granulating will benefit from collagen-based dressings as they lower protease activity64, absorb exudate and promote collagen deposition, whilst still maintaining a moist environment. Hydrocolloid-based dressings are useful for healing PG ulcers that have overlying eschar formation as they stimulate enzymatic degradation of the eschar and this enables for effective re-epithelisation within the wound bed64.
PG wounds are also often suspectable to secondary colonisation and infection, mainly from bacteria, including Staphylococcus aureus, coagulase negative staphylococci and Peptostreptococcus species65. If this occurs, antimicrobial dressings including those containing silver, will reduce the bacterial load due to its ability to damage the bacterial cell wall and membrane64. Topical antibiotics should generally be avoided to prevent the development of resistance.
PG ulcers on the lower legs may lead to the development of oedema secondary to ongoing inflammation. In such cases, gentle compression stockings or wraps, along with leg elevation, may reduce the oedema64.
Conclusion
To date, PG remains a difficult condition to diagnose and treat. The development of diagnostic criteria, including Delphi and PARACELSUS, may assist clinicians in more effectively identifying this condition. Whilst significant advances have been made in the pathophysiology underlying PG, there is a need to further characterise molecular events that occur prior and during the early development of lesions. With regards to treatment, biological therapies trialled in a minority of PG patients, such as IL‑23 and IL‑17 antagonists, have shown promise and may be useful in patients who are not responding to conventional therapy. Clinical translational trials are needed in the future in order to determine whether suppression of the Th17 axis will lead to a downregulation of genes associated with inflammatory pathways, ideally by comparing pre- and post-treatment perilesional samples in PG patients.
Acknowledgements
AF received support from the Australian Government Research Training Program Scholarship.
Conflict of interest
JWF has conducted advisory work for Janssen, Boehringer-Ingelheim, Pfizer, Kyowa Kirin, LEO Pharms, Regeneron and UCB, participated in trials for UCB, Pfizer and Eli Lilly, and received research support from Ortho Dermatologics.
Ethics statement
An ethics statement is not applicable.
Funding
The authors received no funding for this study.
Author(s)
Akshay Flora and John W Frew*
Department of Dermatology, Liverpool Hospital, Sydney, NSW, Australia
Laboratory of Translational Cutaneous Medicine, Ingham Institute of Applied Medical Research, Sydney, NSW, Australia
University of New South Wales, Sydney, NSW, Australia
*Corresponding author Email john.frew@unsw.edu.au
References
- Monari P, Moro R, Motolese A, Misciali C, Baraldi C, Fanti PA, et al. Epidemiology of pyoderma gangrenosum: results from an Italian prospective multicentre study. Int Wound J 2018;15(6):875–9.
- Hobbs MM, Ortega-Loayza AG. Pyoderma gangrenosum: from historical perspectives to emerging investigations. Int Wound J 2020;17(5):1255–65.
- Vignon-Pennamen MD. The extracutaneous involvement in the neutrophilic dermatoses. Clin Dermatol 2000;18(3):339–47.
- Su WP, Davis MD, Weenig RH, Powell FC, Perry HO. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol 2004;43(11):790–800.
- Jorizzo J. Behcet’s disease. J Eur Acad Dermatol Venereol 1997;1001(9):S55.
- George C, Deroide F, Rustin M. Pyoderma gangrenosum – a guide to diagnosis and management. Clin Med (Lond) 2019;19(3):224–8.
- Shankar S, Sterling JC, Rytina E. Pustular pyoderma gangrenosum. Clin Experiment Dermatol 2003;28(6):600–3.
- Edwards FC, Truelove SC. The course and prognosis of ulcerative colitis. Gut 1963;4(4):299–315.
- Bhat R. Pyoderma gangrenosum: an update. Indian Dermatol Online J 2012;3(1):7–13.
- Lear JT, Atherton MT, Byrne JP. Neutrophilic dermatoses: pyoderma gangrenosum and Sweet’s syndrome. Postgrad Med J 1997;73(856):65–8.
- Ambooken B, Khader A, Muhammed K, Rajan U, Snigdha O. Malignant pyoderma gangrenosum eroding the parotid gland successfully treated with dexamethasone pulse therapy. Int J Dermatol 2014;53(12):1536–8.
- Ashchyan HJ, Butler DC, Nelson CA, Noe MH, Tsiaras WG, Lockwood SJ, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol 2018;154(4):409–13.
- Weizman A, Huang B, Berel D, Targan SR, Dubinsky M, Fleshner P, et al. Clinical, serologic, and genetic factors associated with pyoderma gangrenosum and Erythema nodosum in inflammatory bowel disease patients. Inflam Bowel Dis 2014;20(3):525–33.
- Kinchen J, Chen HH, Parikh K, Antanaviciute A, Jagielowicz M, Fawkner-Corbett D, et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell 2018;175(2):372–86.e17.
- Zhang H, Nie X, Shi X, Zhao J, Chen Y, Yao Q, et al. Regulatory mechanisms of the Wnt/乒-Catenin pathway in diabetic cutaneous ulcers. Front Pharmacol 2018;9:1114.
- McBride JD, Jenkins AJ, Liu X, Zhang B, Lee K, Berry WL, et al. Elevated circulation levels of an antiangiogenic SERPIN in patients with diabetic microvascular complications impair wound healing through suppression of Wnt signaling. J Invest Dermatol 2014;134(6):1725–34.
- You HR, Ju JK, Yun SJ, Lee JB, Kim SJ, Won YH, et al. Paraneoplastic pyoderma gangrenosum associated with rectal adenocarcinoma. Ann Dermatol 2018;30(1):79–82.
- Wesolow JT. A rare case of pyoderma gangrenosum in a patient with pancreatic neuroendocrine tumor. Cureus 2020;12(12):e12343.
- Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol 2017;18(4):555–62.
- Marzano AV, Fanoni D, Antiga E, Quaglino P, Caproni M, Crosti C, et al. Expression of cytokines, chemokines and other effector molecules in two prototypic autoinflammatory skin diseases, pyoderma gangrenosum and Sweet’s syndrome. Clin Exp Immunol 2014;178(1):48–56.
- Marzano AV, Ortega-Loayza AG, Heath M, Morse D, Genovese G, Cugno M. Mechanisms of inflammation in neutrophil-mediated skin diseases. Front Immunol 2019;10(1059).
- Sanchez GAM, de Jesus AA, Goldbach-Mansky R. Monogenic autoinflammatory diseases: disorders of amplified danger sensing and cytokine dysregulation. Rheum Dis Clin North Am 2013;39(4):701–34.
- Marzano AV, Borghi A, Meroni PL, Cugno M. Pyoderma gangrenosum and its syndromic forms: evidence for a link with autoinflammation. Br J Dermatol 2016;175(5):882–91.
- Sonbol H, Duchatelet S, Miskinyte S, Bonsang B, Hovnanian A, Misery L. PASH syndrome (pyoderma gangrenosum, acne and hidradenitis suppurativa): a disease with genetic heterogeneity. Br J Dermatol 2018;178(1):e17–e8.
- Weenig RH, Davis MDP, Dahl PR, Su WPD. Skin ulcers misdiagnosed as pyoderma gangrenosum. New Eng J Med 2002;347(18):1412–8.
- Maverakis E, Ma C, Shinkai K, Fiorentino D, Callen JP, Wollina U, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a delphi consensus of international experts. JAMA Dermatol (Chicago, Ill) 2018;154(4):461–6.
- Haag C, Hansen T, Hajar T, Latour E, Keller J, Shinkai K, et al. Comparison of three diagnostic frameworks for pyoderma gangrenosum. J Invest Dermatol 2021;141(1):59–63.
- Binus AM, Qureshi AA, Li VW, Winterfield LS. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol 2011;165(6):1244–50.
- Ting S, Tam M, Kelly R. Scarring in pyoderma gangrenosum. Aust J Dermatol 2021:ajd.13701-undefined.
- Jockenhöfer F, Wollina U, Salva KA, Benson S, Dissemond J. The PARACELSUS score: a novel diagnostic tool for pyoderma gangrenosum. Br J Dermatol (1951) 2019;180(3):615–20.
- González-Moreno J, Ruíz-Ruigomez M, Callejas Rubio JL, Ríos Fernández R, Ortego Centeno N. Pyoderma gangrenosum and systemic lupus erythematosus: a report of five cases and review of the literature. Lupus 2015;24(2):130–7.
- Bernard P, Amici JM, Catanzano G, Cardinaud F, Fayol J, Bonnetblanc JM. Pyoderma gangrenosum and vasculitis. Pathogenic discussion apropos of 3 cases. Ann Dermatol Venereol 1987;114(10):1229–34.
- Wang EA, Steel A, Luxardi G, Mitra A, Patel F, Cheng MY, et al. Classic ulcerative pyoderma gangrenosum is a T cell-mediated disease targeting follicular adnexal structures: a hypothesis based on molecular and clinicopathologic studies. Front Immunol 2017;8:1980.
- Braswell SFB, Kostopoulos TCMD, Ortega-Loayza AGMD. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol 2015;73(4):691–8.
- Mistry P, Carmona-Rivera C, Ombrello AK, Hoffmann P, Seto NL, Jones A, et al. Dysregulated neutrophil responses and neutrophil extracellular trap formation and degradation in PAPA syndrome. Ann Rheum Dis 2018;77(12):1825–33.
- Bonnekoh H, Scheffel J, Wu J, Hoffmann S, Maurer M, Krause K. Skin and systemic inflammation in Schnitzler’s syndrome are associated with neutrophil extracellular trap formation. Front Immunol 2019;10(546).
- Byrd AS, Carmona-Rivera C, O’Neil LJ, Carlucci PM, Cisar C, Rosenberg AZ, et al. Neutrophil extracellular traps, B cells, and type I interferons contribute to immune dysregulation in hidradenitis suppurativa. Sci Transl Med 2019;11(508).
- Marzano AV, Cugno M, Trevisan V, Fanoni D, Venegoni L, Berti E, et al. Role of inflammatory cells, cytokines and matrix metalloproteinases in neutrophil-mediated skin diseases. Clin Exp Immunol 2010;162(1):100–7.
- Marzano AV, Damiani G, Ceccherini I, Berti E, Gattorno M, Cugno M. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis). Br J Dermatol 2017;176(6):1588–98.
- Marzano AV, Ceccherini I, Gattorno M, Fanoni D, Caroli F, Rusmini M, et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine (Baltimore) 2014;93(27):e187.
- Kanni T, Zenker O, Habel M, Riedemann N, Giamarellos-Bourboulis EJ. Complement activation in hidradenitis suppurativa: a new pathway of pathogenesis? Br J Dermatol 2018;179(2):413–9.
- Bister V, Mäkitalo L, Jeskanen L, Saarialho-Kere U. Expression of MMP-9, MMP-10 and TNF-alpha and lack of epithelial MMP-1 and MMP-26 characterize pyoderma gangrenosum. J Cutan Pathol 2007;34(12):889–98.
- Baier C, Barak O. Guselkumab as a treatment option for recalcitrant pyoderma gangrenosum. JAAD Case Rep 2021;8:43–6.
- Guenova E, Teske A, Fehrenbacher B, Hoerber S, Adamczyk A, Schaller M, et al. Interleukin 23 expression in pyoderma gangrenosum and targeted therapy with ustekinumab. Arch Dermatol 2011;147(10):1203–5.
- Tee MW, Avarbock AB, Ungar J, Frew JW. Rapid resolution of pyoderma gangrenosum with brodalumab therapy. JAAD Case Rep 2020;6(11):1167–9.
- Clark RA, Schlapbach C. T(H)9 cells in skin disorders. Semin Immunopathol 2017;39(1):47–54.
- Antiga E, Maglie R, Volpi W, Bianchi B, Berti E, Marzano AV, et al. T helper type 1-related molecules as well as interleukin-15 are hyperexpressed in the skin lesions of patients with pyoderma gangrenosum. Clin Exp Immunol 2017;189(3):383–91.
- Caproni M, Antiga E, Volpi W, Verdelli A, Venegoni L, Quaglino P, et al. The Treg/Th17 cell ratio is reduced in the skin lesions of patients with pyoderma gangrenosum. Br J Dermatol 2015;173(1):275–8.
- Ortega-Loayza AG, Friedman MA, Reese AM, Liu Y, Greiling TM, Cassidy PB, et al. Molecular and cellular characterization of pyoderma gangrenosum: implications for the use of gene expression. J Invest Dermatol 2021.
- Herberger K, Dissemond J, Hohaus K, Schaller J, Anastasiadou Z, Augustin M. Treatment of pyoderma gangrenosum: retrospective multicentre analysis of 121 patients. Br J Dermatol (1951) 2016;175(5):1070–2.
- Wenzel J, Gerdsen R, Phillipp-Dormston W, Bieber T, Uerlich M. Topical Treatment of pyoderma gangraenosum. Dermatol 2002;205(3):221–3.
- Richter-Hintz D, Schuppe H-C, Homey B, Lehmann P, Ruzicka T. Topical tacrolimus (FK 506) is effective in the treatment of pyoderma gangrenosum. J Am Acad Dermatol 2000;42(2):304.
- Chiba T, Isomura I, Suzuki A, Morita A. Topical tacrolimus therapy for pyoderma gangrenosum. J Dermatol 2005;32(3):199–203.
- Thomson AW, Bonham CA, Zeevi A. Mode of action of tacrolimus (FK506): molecular and cellular mechanisms. Ther Drug Monit 1995;17(6):584–91.
- Lyon CC, Stapleton M, Smith AJ, Mendelsohn S, Beck MH, Griffiths CE. Topical tacrolimus in the management of peristomal pyoderma gangrenosum. J Dermatolog Treat 2001;12(1):13–7.
- Hughes AP, Jackson JM, Callen JP. Clinical features and treatment of peristomal pyoderma gangrenosum. JAMA 2000;284(12):1546–8.
- Ormerod AD, Thomas KS, Craig FE, Mitchell E, Greenlaw N, Norrie J, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ 2015;350:h2958.
- Johnson RB, Lazarus GS. Pulse therapy: therapeutic efficacy in the treatment of pyoderma gangrenosum. Arch Dermatol 1982;118(2):76–84.
- Brooklyn TN, Dunnill MGS, Shetty A, Bowden JJ, Williams JDL, Griffiths CEM, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut 2006;55(4):505–9.
- Yamasaki K, Yamanaka K, Zhao Y, Iwano S, Takei K, Suzuki K, et al. Adalimumab in Japanese patients with active ulcers of pyoderma gangrenosum: twenty-six-week phase 3 open-label study. J Dermatol 2020;47(12):1383–90.
- Charles CA, Leon A, Banta MR, Kirsner RS. Etanercept for the treatment of refractory pyoderma gangrenosum: a brief series. Int J Dermatol 2007;46(10):1095–9.
- Kochar B, Herfarth N, Mamie C, Navarini AA, Scharl M, Herfarth HH. Tofacitinib for the treatment of pyoderma gangrenosum. Clin Gastroenterol Hepatol 2019;17(5):991–3.
- Schultz GS, Sibbald RG, Falanga V, Ayello EA, Dowsett C, Harding K, et al. Wound bed preparation: a systematic approach to wound management. Wound Repair Regen 2003;11 Suppl 1:S1–28.
- Croitoru D, Naderi-Azad S, Sachdeva M, Piguet V, Alavi A. A wound care specialist’s approach to pyoderma gangrenosum. Adv Wound Care (New Rochelle, NY) 2020;9(12):686–94.
- Bowler PG, Duerden BI, Armstrong DG. Wound microbiology and associated approaches to wound management. Clin Microbiol Rev 2001;14(2):244–69.
- Ahronowitz I, Harp J, Shinkai K. Etiology and management of pyoderma gangrenosum: a comprehensive review. Am J Clin Dermatol 2012;13(3):191–211.

