

Volume 30 Number 1
Alopecia areata: a review of diagnosis, pathogenesis and the therapeutic landscape
Anthony Moussa, Laita Bokhari and Rodney D Sinclair
Keywords alopecia areata, alopecia totalis, alopecia universalis, hair disorder
For referencing Moussa A et al. Alopecia areata: a review of diagnosis, pathogenesis and the therapeutic landscape. Wound Practice and Research 2022; 30(1):24-33.
DOI
https://doi.org/10.33235/wpr.30.1.24-33
Submitted 18 January 2022
Accepted 8 February 2022
Abstract
Alopecia areata (AA) is a T cell-mediated autoimmune disease that has an estimated lifetime prevalence of 2.1%. Although clinically heterogenous, the typical presentation is of discrete, well-demarcated patches of non-scarring hair loss. In severe AA, complete loss of scalp hair (alopecia totalis, AT) or all hair (alopecia universalis, AU) may occur. There is considerable variation in how AA is treated by dermatologists. Recent insights into the aetiopathogenesis of AA have led to the emergence of several novel treatment strategies, including Janus kinase inhibitors and biologics. This review provides a contemporary evidence-based summary of AA, including clinical features, severity assessment, disease pathogenesis and current and emerging treatment strategies.
Introduction
Alopecia areata (AA) is a relapsing and remitting T cell-mediated autoimmune disease characterised by variable degrees of circular, non-scarring patches of hair loss1. AA can occur at any age in genetically susceptible individuals following unknown environmental triggers and has an estimated lifetime prevalence of 2.1%2. While any hair-bearing area may be involved, the scalp is most commonly affected2,3. Health-related quality of life may be significantly compromised, with patients at an increased risk of psychiatric comorbidities, including social phobia, anxiety, depression and suicidal ideation4,5.
AA is clinically heterogenous and has an unpredictable clinical course. In severe cases, patients may suffer total loss of scalp hair (alopecia totalis, AT) or total loss of all hair on the body (alopecia universalis, AU)1,3. Sufferers are said to have acute AA if hair loss is followed by spontaneous regrowth within 12 months or chronic AA if hair loss persists beyond 12 months. One third of patients have chronic AA; of those, 45% will progress to AT or AU6. Factors associated with poor prognosis and increased likelihood of refractory disease include extensive hair loss (>50% of the scalp), an ophiasis distribution, comorbid atopic disease, earlier age of onset and nail involvement7. Patients with AA are also at an overall increased risk of other autoimmune diseases and atopic conditions1,8. Collapse of hair follicle immune privilege (IP) has been implicated in AA pathogenesis9,10. The treatment of AA aims to arrest disease progression and reverse hair loss. In this review article we summarise the clinical features of AA, explore disease pathogenesis, and discuss current and novel therapeutic approaches.
Epidemiology
AA has a lifetime incidence ranging between 0.7–4%2,11. While individuals of all ages may be affected, 82.6–88% of sufferers experience their first episode by the age of 40 years12. In paediatric patients under the age of 16 years, the mean age of diagnosis is 11.2 years13. Although no gender predilection is reported2, a cross-sectional study by Lee et al. looking at a US population noted higher odds of AA in African Americans and lower odds in Asians compared with whites14. Furthermore, a family history of AA is reported in up to 8.6% of adult patients and in 10–51.6% of paediatric patients12. A diagnosis of AA confers an increased risk of other autoimmune diseases, including autoimmune thyroid disease, systemic lupus erythematosus, vitiligo, inflammatory bowel disease, psoriasis and rheumatoid arthritis1. Co-morbid atopic disease is seen in 39% of AA patients8.
Clinical features
AA is a clinically heterogenous condition and may manifest in several different patterns. The typical presentation is of a discrete, well-demarcated, non-scarring patch of hair loss (Figure 1). The skin is smooth (“like a baby’s bottom”) and may occasionally be slightly erythematous, but without any epidermal change3,15. In active disease, a pull test at the margin of an AA lesion is positive and produces telogen or dystrophic anagen hairs1,15. The ensuing clinical course is unpredictable. The initial patch may expand over weeks to months or additional patches may develop at varying intervals. Indeed, the presence of a solitary patch, the most common initial presentation, may increase the risk of developing further patches. New patches may subsequently merge to form larger patches1,3,15. Hair loss is typically asymptomatic; however, some patients report a prodromal localised dysesthesia prior to onset of an AA lesion15.
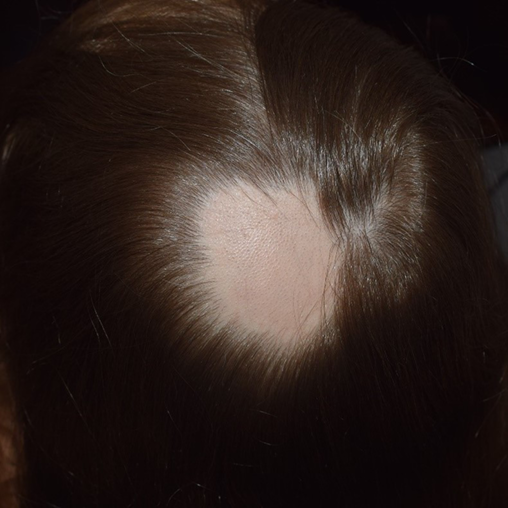
Figure 1. Example of acute patch of AA affecting the occiput
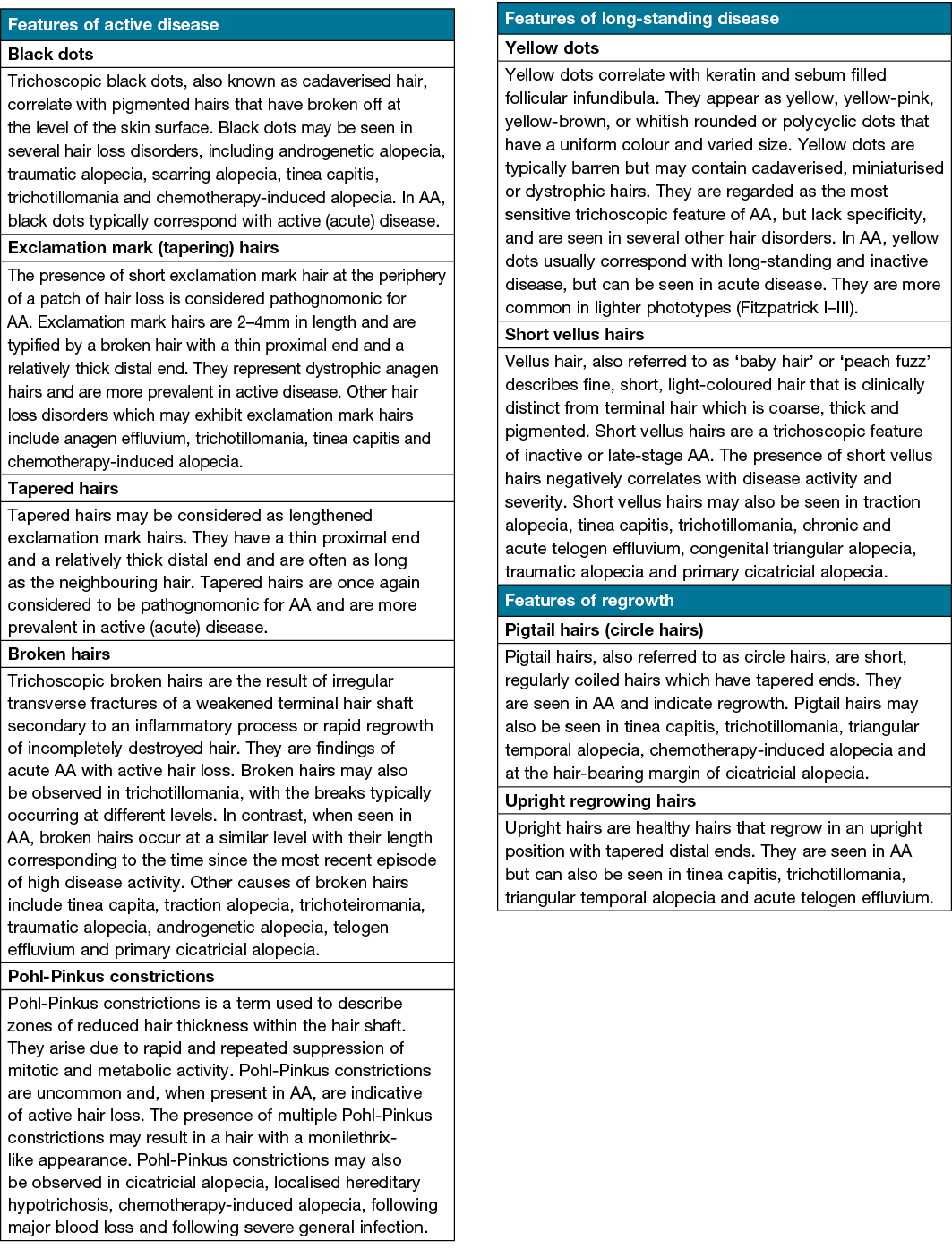
Trichoscopy, also known as hair and scalp dermatoscopy, may help to facilitate a diagnosis of AA and monitor disease activity (Table 1). Trichoscopic features of active AA include black dots, exclamation point hairs, tapered hairs and broken hairs. Features of long-standing AA include yellow dots and short vellus hairs16,17.
Table 1. Trichoscopic features of AA16,17
Approximately 40% of patients with AA will only ever develop a solitary patch which, if left untreated, spontaneously regrows within 6 months. An additional 27% of patients will develop further patches (Figures 2 & 3) yet still achieve complete remission at 1 year. However, most patients will relapse in the months or years after remission6,15. Patients with chronic AA, where patches persist beyond 1 year, have a 30% risk of progressing to total loss of scalp hair (AT) and a 15% risk of losing all hair on the body (AU) (Figure 4)6. Hair loss in AA may also occur in an ophiasis distribution, characterised by band-like hair loss extending across the occipital scalp (Figure 5). An inverse ophiasis pattern called sisaipho has also been described; this pattern of AA resembles male androgenetic alopecia and involves loss of hair in the frontal, temporal and parietal scalp with sparing of the scalp periphery. Occasionally patients may present with diffuse AA, often referred to as AA incognita, without any discrete patches (Figure 6)1,3,15.
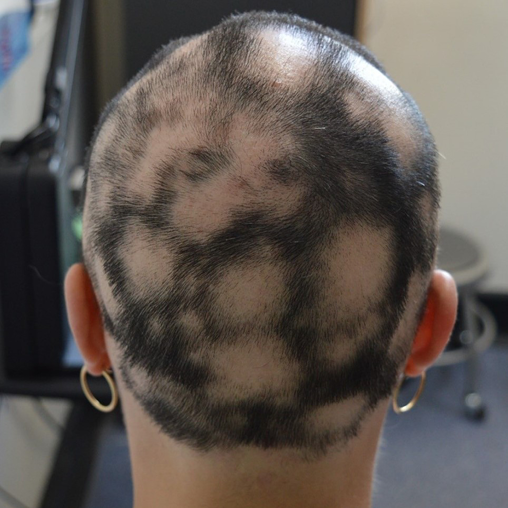
Figure 2. Example of patchy AA
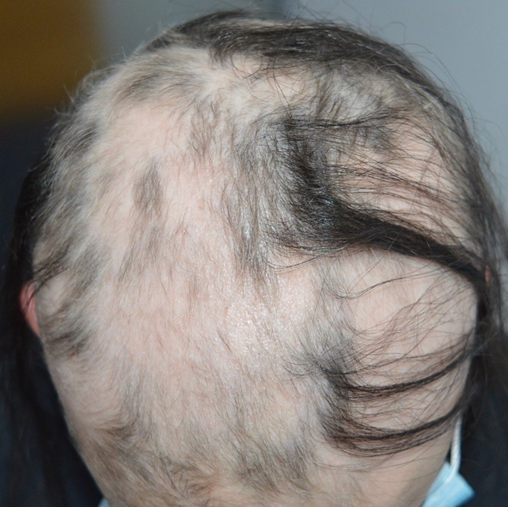
Figure 3. Example of patchy AA with mixed areas of regrowth and coalescing patches
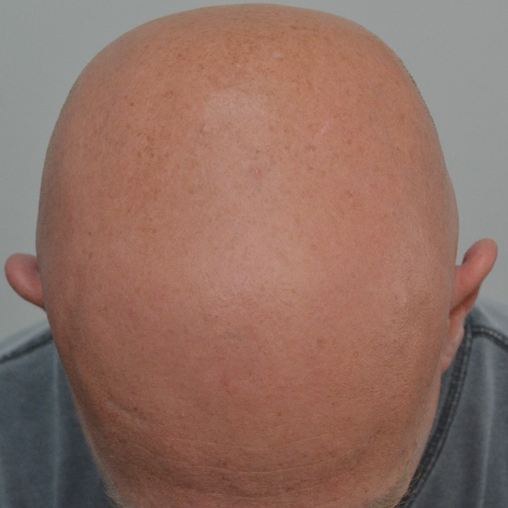
Figure 4. Example of AT
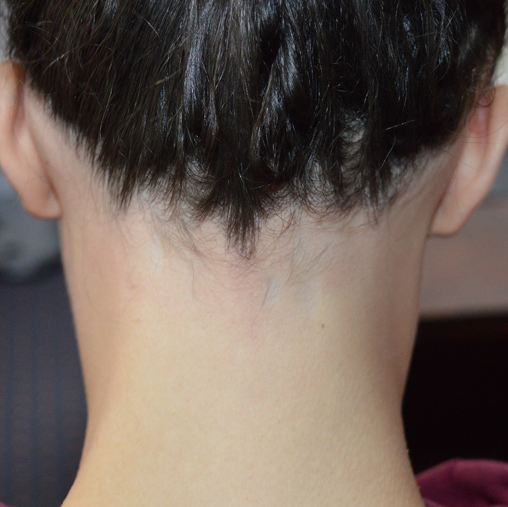
Figure 5. Example of ophiasis type AA
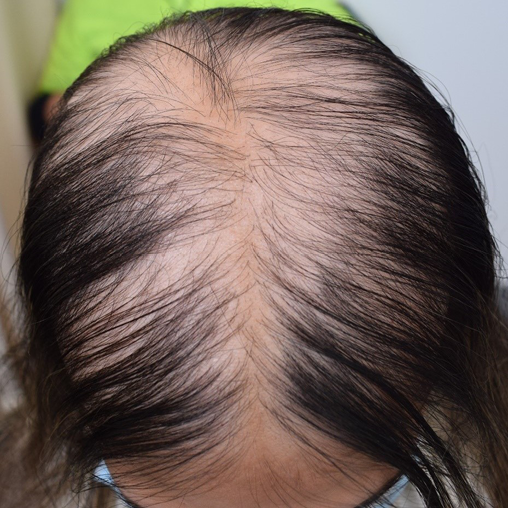
Figure 6. Example of acute diffuse AA
Although AA can affect any hair-bearing site, the scalp is most affected and is often the first to develop a lesion1. Eyebrow and eyelash loss are common, and the beard is affected in 28% of males18. For reasons which are not fully understood, white hairs appear to be relatively spared in AA compared to pigmented hairs. In patients with rapid disease progression, this phenomenon may account for reports of hair “turning white overnight” (canities subita)3,15. Furthermore, it is our experience that AA is uncommon in redheads; indeed, red hair may be protective against AA.
As AA is a non-scarring alopecia (i.e., does not destroy hair follicles), regrowth of hair can occur even many years after hair loss and possibly lifelong. Regrowth occurs within patches with hairs that are usually white and fine but gradually become thicker and re-pigmented (Figure 7). Of note, regrowth and progression of AA can occur simultaneously in separate parts of the scalp1,3,15.
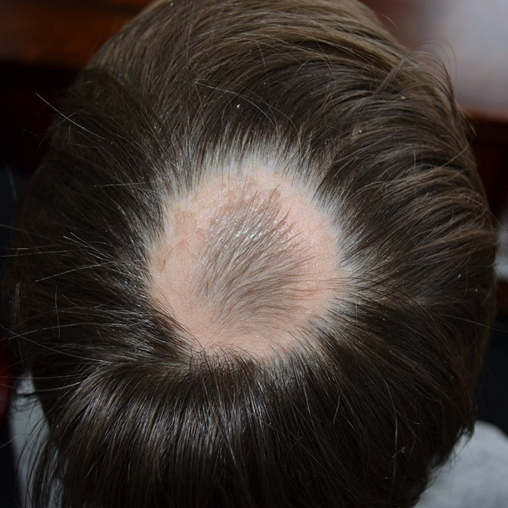
Figure 7. Example of regrowth of hair within an AA patch
Nail disease occurs in 10–15% of cases of AA and may precede, succeed or coincide with active hair loss. Nail involvement may include pitting, onychorrhexis, trachyonychia, punctuate leukonychia, onycholysis, onychomadesis and red spots on the lunulae19. Due to the association with AU and AT, nail involvement may be considered a poor prognostic factor3,7.
Diagnosis
Patient history and physical examination usually render the diagnosis of AA straightforward. For patients in whom the diagnosis remains uncertain, a skin biopsy is usually diagnostic1,3. Histopathological features of AA vary with the acuity of hair loss in the area biopsied20. Acute disease is most commonly typified by a dense, peribulbar, lymphocytic infiltrate surrounding anagen hair follicles that has been described as a “swarm of bees” pattern. Features of subacute disease include a reduction in anagen hairs and an increase in the catagen and telogen hairs. In contrast, chronic disease is characterised by decreased terminal hair and increased miniaturised hair with variable degrees of less pronounced inflammation20,21. In one study of 50 AA patients subjected to histopathological investigation, samples obtained from perilesional, clinically unaffected scalp demonstrated microscopic abnormalities in 66% of cases22. However, the role of histopathological sampling of non-affected scalp for assessment of AA spread and management is not established. Important conditions that may present as differentials for AA include trichotillomania, tinea capitis, traction alopecia, aplasia cutis, temporal triangular alopecia and secondary syphilis1,15.
Severity staging
Historically, AA has been categorised based on the pattern of hair loss as patchy AA, AT, AU, diffuse AA and ophiasis3,15. Despite widespread use, such terms are only vaguely descriptive. Several tools have been created to assist with assessing scalp AA severity in clinical practice and clinical trials23–31.
Severity of Alopecia Tool (SALT) scoring
The Severity of Alopecia Tool (SALT) scoring method is the most widely used tool in clinical practice and clinical trials to assess AA severity and monitor response to treatment. First proposed by Olsen et al. in 2004, the SALT score divides the scalp into four affected regions: the top (40%), back (24%) and left and right sides (18% each). A score of 0 indicates a full head of hair while a score of 100 corresponds with complete loss of hair24. In practice, the SALT score is quick and easy to perform. However, despite its widespread use, the SALT score does not consider non-scalp factors which may modulate disease severity such as facial hair involvement or psychosocial morbidity. Furthermore, calculating the SALT score is less useful when subtle changes of hair loss predominate. Olsen et al. subsequently proposed the SALT II visual aid in 2016 which more finely divides the assessment of scalp hair loss and permits the assessor to accurately define concomitant areas of non-AA hair loss25.
Alopecia Areata Investigator Global Assessment (AA-IGA™) Scale
The AA Investigator Global Assessment (AA-IGA™) Scale uses the SALT score to denote disease severity based on discrete SALT ranges26. It measures five clinically meaningful graduations of scalp hair loss as defined by clinician and patient perspectives. Disease severity is defined based on the following percentage of scalp hair loss – none (0%), limited (1–20%), moderate (21–49%), severe (50–94%) and very severe (95–100%)26. Like the SALT score, the AA-IGA does not consider non-scalp related hair loss or psychosocial burden. Furthermore, the ranges used in the AA-IGA are not universally accepted by all dermatologists.
Alopecia Density and Extent (ALODEX) score
The Alopecia Density and Extent (ALODEX) score is a method that combines both extent and density of hair loss to produce the final percentage of hair loss. It is based on the SALT II visual aid and, while possible to calculate manually, is most rapidly performed electronically using an iPad application23.
Alopecia Areata Progression Index (AAPI)
The Alopecia Areata Progression Index (AAPI) was developed by Jang et al. in 2016. Like calculation of the SALT score, the scalp is divided into four quadrants. However, in addition to percentage scalp hair loss, AA disease activity is assessed using a hair pull test and trichoscopic findings27. Once again, non-scalp related hair loss or psychosocial burden is not considered using this scoring tool.
Alopecia Areata Severity Index (AASI)
The Alopecia Areata Severity Index (AASI) is a relatively new tool described by Majid et al. in a 2021 pilot study. It entails individual assessment of the scalp, upper face and beard for hair loss in order to produce a total severity score (i.e., AASI = AASIscalp + AASIbeard + AASIupper face). The maximum AASI score is 250 and 150 for males and females respectively28. However, it has only been tested on a single population group and requires further validation.
Alopecia Areata Scale
The Alopecia Areata Scale has been recently proposed following an academic–industry collaborative of 22 dermatologists experienced in the management of AA. Based on the extent of scalp hair loss, AA severity is defined as mild (≤20%), moderate (21–49%) or severe (50–100%)29. In patients with mild or moderate disease, AA severity rating is increased by one level if any of the following are present:
- Negative impact on psychosocial functioning resulting from AA.
- Noticeable involvement of eyebrows or eyelashes.
- Inadequate response after at least 6 months of treatment.
- Diffuse (multifocal) positive hair pull test consistent with rapidly progressive AA29.
Non-scalp measures of AA severity
Separate measures of non-scalp AA severity have been developed for use in clinical trials and include the Clinician-Reported Outcome (ClinRO) and Patient-Reported Outcome (PRO) Measures for Eyebrow, Eyelash and Nail Assessment30. Stefanis et al. in 2021 proposed a novel scoring system to estimate the extent of beard hair loss in AA, the Alopecia Barbae Severity score (ALBAS score); however, this requires further validation31.
Assessing psychosocial burden
While lacking in most AA severity assessment tools, psychosocial burden in AA has been separately assessed using tools such as the Hospital Anxiety and Depression Scale (HADS), the Dermatology Life Quality Index (DLQI) and the Perceived Stress Scale (PSS-14)23.
Pathogenesis
AA is a cytotoxic T cell-mediated autoimmune disease of largely unknown pathogenesis. The prevailing theory centres upon loss of hair follicle IP in genetically predisposed individuals following unknown environmental triggers9,10.
Genetics
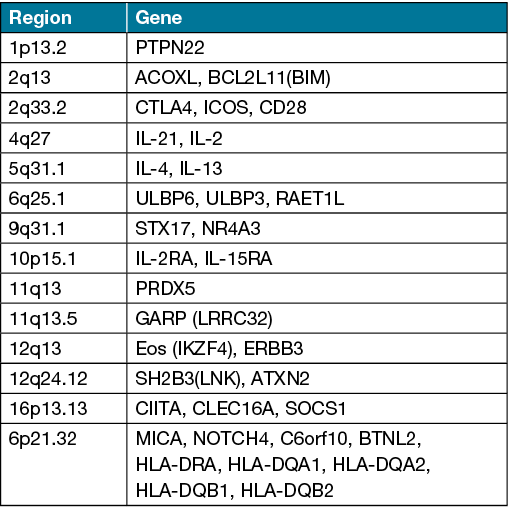
The role of genetic factors in the development of AA is well documented. Observational studies have demonstrated a heightened risk of disease in individuals with a first-degree relative compared to the general population (8.6–51.6% vs 2.1%)2,12. Genome-wide association studies have implicated 14 genomic loci in AA containing genes related to regulators of T cell function such as cytotoxic T lymphocyte-associated antigen 4 and interleukin (IL)‑2/IL‑21, and natural killer cell receptor (NKG2D) activating ligands including MHC class I chain-related protein A (MICA) and UL16 Binding Protein (ULBP)‑3/6 (Table 2)32,33. While a large number of genes have been identified, no monogenic cause of AA has been isolated to date. A 2015 genome-wide meta-analysis in AA by Betz et al. implicated human leukocyte antigen‑DR (HLA‑DR) on chromosome 6 as the key aetiological driver33. A study by Fischer et al. investigating copy number variants identified duplications spanning the genes melanin concentrating hormone receptor 2 (MCHR2) and MCHR2 Antisense RNA 1, implicated in melanin-concentrating hormone (MCH) signalling34. MCHR2 has been previously shown to affect the scale colour of barfin flounder fish via the induction of melanin aggregation. As pigmented hair is preferentially affected in AA, this may suggest a relationship between pigmentation, MCH signalling and AA34. Interestingly, filaggrin gene mutations have also been detected in patients with concomitant AA and atopic dermatitis (AD) and may be associated with AA severity35. Furthermore, the concordance rate of AA in monozygotic twins is 42–55%, suggesting both genetic and environmental triggers12.
Table 2. Genes with significant association in AA genome-wide association studies32,33
Collapse of hair follicle IP and cytokines
Specific sites within the body can tolerate the introduction of antigens without the development of an immune response, a concept referred to as immune privilege (IP). In health, anagen hair follicle outer root sheath cells fail to express class 1 major histocompatibility antigens (MHC‑1) and exhibit relative IP from the level of the bulge to the bulb9,10. Although preservation of IP and formation of a local immunoinhibitory milieu is mainly achieved via downregulation of MHC‑1 in anagen hair bulbs, production of local immunosuppressant molecules such as α‑melanocyte‑stimulating hormone (α‑MSH), transforming growth factor‑β (TGF‑β), vasoactive intestinal peptide (VIP) and indoleamine‑2,3‑dioxygenase (IDO) also contribute9. Ultimately, IP protects the anagen follicle from premature catagen10.
Recent mouse studies have shed light on the role of interferon (IFN)‑γ producing cytotoxic CD8+NKG2D+ effector T cells in AA pathogenesis. In the normal state, senescent cells in the anagen hair bulb express cell surface NKG2D activating ligands such as ULBP3, ULBP6 and MICA9,10. Activation of IFN‑γ producing cytotoxic CD8+NKG2D+ effector T cells requires co‑expression of these NKG2D activating ligands together with MHC‑1 antigens10. Collapse of hair follicle IP is associated with hair bulb outer root sheath keratinocyte MHC‑1 expression resulting in CD8+NKG2D+ effector T cells binding to damaged hair bulb. Binding of CD8+NKG2D+ T cells to hair follicle bulb keratinocytes results in the release of IFN‑γ which binds to its receptor on hair follicle epithelial cells; this induces catagen and promotes the production of interleukin (IL)‑15 via Janus kinase (JAK)‑1 and JAK‑2. Subsequently, IL‑15 binds to its receptor on CD8+NKG2D+ T cells and induces JAK‑1 and JAK‑3‑mediated IFN‑γ production, thereby completing the feedback loop. Premature termination of the anagen phase results in hair loss9,10.
In addition to T‑helper type 1 (Th1) cells, a contributory role of T‑helper 17 (Th17) cells and T regulatory (Treg) cells has been suggested in the pathogenesis of AA36–38. Immunofluorescent staining of scalp samples from patients with AA has demonstrated infiltration of CD4+IL‑17A+ Th17 cells in the dermis, particularly around hair follicles36. Furthermore, several studies have revealed an increase in serum and lesion Th17‑related cytokine levels, including IL‑17 and IL‑22 in patients with AA37,38. In a study by El‑Morsi et al, serum IL‑17A levels demonstrated significant negative correlation to age of onset37. A separate study by Loh et al. demonstrated a positive correlation with serum IL‑17 levels and AA severity; however, interestingly, IL‑17 levels were decreased in patients with acute diffuse total alopecia (ADTA) subsets38. Serum levels of the Treg cytokines, TGF‑β and IL‑10 were also significantly increased in AA. As IL‑1β and TGF‑β promote Th17 differentiation, this led the authors to suggest an enhanced propensity of Treg cells to differentiate into IL‑17A‑producing cells in AA38.
While AA is typically regarded as a type 1 inflammatory disease (IFN‑γ is the key cytokine), 15.6% of adults and 39.5% of children with AA have co‑morbid AD, a type 2 inflammatory disease10,12. Furthermore, the presence of AD is regarded as a poor prognostic marker in AA7,35. Despite this, the immunological relationship between the two conditions is poorly understood. AD is principally a Th‑2 driven disease, typified by elevated levels of IL‑4, IL‑5, IL‑13 and IL‑3139. Dupilumab is a human monoclonal antibody directed against the α subunit of the IL‑4 receptor that is shared by IL‑4 and IL‑13. Dupilumab has shown dual efficacy in the management of AA and AD; however, cases of AA development following dupilumab have also been reported40. AD has recently been divided into extrinsic and intrinsic types. Extrinsic AD is typified by high expression of type 2 cytokines (IL‑4, IL‑5 and IL‑13), high serum levels of IgE and skin barrier dysfunction. In contrast, intrinsic AD is characterised by high expression of IFN‑γ, low expression of type 2 cytokines, normal serum IgE levels and normal skin barrier function39. The differences between extrinsic and intrinsic AD may account for the different trajectories observed following treatment of co‑morbid AA with dupilumab. In cases of dupilumab-induced AA, disruption of IL‑4 and IL‑13 processes in Th2‑mediated inflammation may provoke a Th1‑shift in the immune milieu39.
Management
There is significant variation in the way AA is treated between dermatologists. To date, several guidelines and expert consensus statements have been proposed to aid with the management of disease6,41,42. Holistic management requires consideration of a sufferer’s psychosocial disease burden. Recent advances into the aetiopathogenesis of AA have led to the development of several novel targeted treatment strategies.
Conservative measures
As up to 80% of patients with acute and limited AA may undergo spontaneous remission, observation and reassurance alone may be appropriate management for some6,41. Additional camouflage techniques that have been used with success include wigs, colour‑matched wool fibres, hair extensions and top pieces6.
Topical therapy
Topical treatment may also be considered in limited to moderate disease to accelerate hair regrowth. Topical treatments include corticosteroids, minoxidil and immunotherapy6,41–43. Several strategies have been proposed in the British Association of Dermatologists (BAD) and Japanese Dermatological Association (JDA) guidelines for the management of AA, and in an Australian expert consensus statement6,41,42.
Topical corticosteroids
Potent topical corticosteroids are traditionally used as first‑line therapy in children and in adults unable to tolerate intralesional corticosteroids6. They are thought to reduce and contain inflammation in AA lesions and to accelerate recovery of damaged hair follicles. Despite widespread use, there are conflicting reports of efficacy41,42. Long-term use is best avoided; however, despite concerns, skin atrophy is rarely seen6.
Topical immunotherapy
Topical immunotherapy has been used to treat AA since 197641. Immunotherapeutic agents such as diphenylcyclopropenone (DPCP) and squaric acid dibutyl ester (SADBE) are thought to work via induction of antigenic competition which distracts CD4+ T cells from attacking hair follicles. A high response rate (88–100%) has been reported in patchy AA; however, this is significantly less in severe AA (60%) and AT/AU (17%)43. Topical immunotherapy is recommended for moderate to severe AA of all ages by the JDA guidelines42. The Australian expert consensus guidelines suggest using topical immunotherapy as a second-line therapy or as first‑line therapy in patients with extensive disease, in ophiasis pattern AA, and in children6. Treatment can be time consuming and adverse effects may include blistering, urticaria and depigmentation41,43.
Dithranol
Topical dithranol is another second‑line treatment option in patients with limited or extensive disease. Treatment aims to induce a low‑grade, irritant dermatitis. Despite some reports of efficacy, topical dithranol appears to be less effective than topical immunotherapy and intralesional corticosteroids. Irritation and staining of the skin may also be problematic adverse effects41.
Topical minoxidil
Minoxidil, a hair growth promoter, has been used in the treatment of various hair loss disorders, including androgenetic alopecia, and appears to demonstrate a dose response effect6,41. Cosmetically acceptable regrowth has been observed in cases of limited AA; however, it is cosmetically ineffective in AT and AU. If used, topical minoxidil should be combined as adjunctive treatment. Although generally well tolerated, facial hypertrichosis and dermatitis are possible adverse events6,41.
Intralesional corticosteroids
Intralesional corticosteroids, such as long‑acting triamcinolone acetonide, are routinely used to treat adults with isolated patches of AA (<50% scalp involvement), and are recommended as a first-line therapy in adults in the JDA guidelines and Australian expert consensus statement6,42. Triamcinolone is usually mixed with lignocaine or normal saline. Concentrations of 5mg/mL are used for the scalp and 2.5mg/mL are used for the eyebrows, face and beard. Injections are repeated every 4–6 weeks until regrowth occurs and discontinued at 6 months if no regrowth is observed6. Intralesional corticosteroids typically demonstrate better results than topical corticosteroids but are less suitable in children under the age of 10. Potential risks include skin atrophy and hypopigmentation (particularly in darker phototypes)6,41.
Systemic corticosteroids
Oral corticosteroids may be prescribed to arrest rapidly progressing and extensive hair loss in AA and in refractory cases. Although therapy is often effective in up to 80% of patients, AA relapse may be seen in 50% of responders upon dose reduction or following discontinuation of treatment6,41. Several dosing strategies may be implemented. Prednisolone may be commenced at a high dose (0.5–0.75mg/kg) and tapered from 6–12 weeks; alternatively, patients may be commenced and maintained on a static dose (0.25mg/kg) for 6–12 weeks or commenced on a lower dose (0.1–0.2mg/kg) that is up-titrated according to response and tolerability6. Oral corticosteroids may also be prescribed as pulse therapy42. The potential risks of protracted oral corticosteroid use limit the duration of treatment6,41. The Australian expert consensus statement guidelines suggest switching to a steroid-sparing agent in patients who are corticosteroid responsive, but also corticosteroid dependent6. The JDA guidelines echo this sentiment and recommend limiting oral corticosteroid use to adults with rapidly progressive AA of ≥25% scalp involvement42. An alternative to oral ingestion is the intramuscular (IM) or intravenous (IV) delivery of long‑acting corticosteroids. There is a suggestion that IM and IV delivery may offer a superior side‑effect profile compared to oral delivery6. IV pulse therapy is widely used in Japan and listed in the JDA guidelines as a treatment option for adults with acute and severe AA; it is not routinely used in Australia, Europe or North America42.
Steroid‑sparing agents
Treatment of severe and recalcitrant AA is challenging. Agents such as methotrexate, azathioprine and cyclosporine have been used as monotherapy or in combination with oral corticosteroids for the management of severe AA6,44.
Methotrexate is a disease‑modifying antirheumatic‑drug (DMARD) that has demonstrated reasonable efficacy in the management of severe AA6,44. Although methotrexate can be used as monotherapy, combination treatment with oral corticosteroids results in a higher complete response rate. Furthermore, compared to children, adults appear to be more responsive to treatment44. Treatment is commonly initiated at a dose of 5–10mg once a week and gradually up-titrated every 4–6 weeks according to response and tolerability (max dose: 20–30mg/week)6. Unfortunately, tapering of methotrexate frequently results in AA recurrence. Furthermore, treatment with methotrexate requires close monitoring given potential side effects and toxicities; concomitant treatment with folic acid supplements is thought to reduce these risks6,44.
Azathioprine is a DMARD that had be used to induce hair regrowth in patients with moderate to severe AA. To minimise the risk of gastrointestinal upset, it is typically initiated at a low dose (0.5–1mg/kg daily) and gradually up-titrated every 4–6 weeks according to response and tolerability (max dose: 2–3mg/kg daily)6.
Cyclosporine is a calcineurin inhibitor that has been used to induce hair regrowth in AA. It is typically initiated at a dose of 2mg/kg daily (in three divided doses) and gradually up‑titrated every 4–6 weeks based on response and tolerability (max dose: 5mg/kg). Cyclosporine has serious potential risks which are increased with longer duration of treatment, including gastrointestinal upset, nephrotoxicity and hepatotoxicity6.
Platelet-rich plasma (PRP)
Although a newer therapeutic modality, platelet‑rich plasma (PRP) exerts anti-inflammatory effects that have been shown to induce regrowth upon injection into AA affected areas in randomised controlled trials42. PRP is derived from whole blood that has been centrifuged to remove red blood cells, leaving a concentrate of PRP protein. Further studies are needed to confirm its efficacy.
Other systemic agents
Several other systemic agents have been described in small, uncontrolled retrospective case series and case reports. Treatments include dapsone, mycophenolate mofetil, ezetimibe‑simvastatin, oral minoxidil, sulfasalazine and psoralen plus ultraviolet A (PUVA) photochemotherapy6,41,42,45.
Emerging therapeutic approaches
Biologics
Several biologics directed against Th1 and Th2 cytokines have been trialled for management of AA. Dupilumab, an IL‑4Rα blocker that inhibits both IL‑4 and IL‑13, has demonstrated success in managing AA in patients with concomitant AD39. However, development of new AA and exacerbation of pre‑existing AA has also been reported with dupilumab use40. Tildrakizumab, a high‑affinity monoclonal antibody targeting IL‑23, was of little benefit in patients with chronic AA46. Similarly, ustekinumab, an IL‑12/IL‑23p40 monoclonal antibody, showed mixed reports, both improving and exacerbating disease47. Furthermore, despite the proposed role of the Th17/IL‑17 axis in AA pathogenesis, several IL‑17 inhibitors, including brodalumab, ixekizumab and secukinumab, have demonstrated no to little benefit in treating AA to date. The TNF‑α inhibitors etanercept and adalimumab have also been shown to be ineffective treatments despite the dominant theory underlying AA pathogenesis47.
Janus kinase (JAK) inhibitors
Janus kinase (JAK) inhibitors have emerged as a promising new treatment option for moderate to severe AA. The JAK family is a group of four intracellular enzymes (JAK1, JAK2, JAK3 and TYK2) involved in immune response, defence and haematopoesis11. To date, pharmacologic inhibition of the JAK enzyme family has been used to treat several immune-mediated diseases, including AD, psoriasis, systemic lupus erythematosus, inflammatory bowel disease and rheumatoid arthritis11. Several JAK inhibitors have demonstrated efficacy in the management of AA (Table 3)11,48–50.
Table 3. JAK inhibitors used to treat AA including those under investigation11,47–50
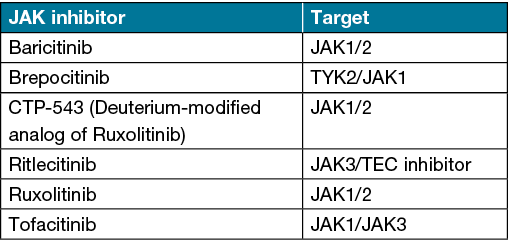
Tofacitinib is a JAK‑1/3 inhibitor that has demonstrated efficacy in several studies for the treatment of adult and paediatric AA11,48. In an open label study by Almutairi et al., AA patients treated with tofacitinib 5mg twice daily demonstrated a mean reduction in SALT score of 95.2% after 6 months48. Similarly, patients treated with ruxolitinib (20mg twice daily), a selective JAK‑1/2 inhibitor, demonstrated a mean reduction in SALT score of 93.8% after 6 months48. Baricitinib, a selective and reversible JAK‑1/2 inhibitor has demonstrated success in treating severe AA in a phase II study of 110 adults49. In this study, the proportion of patients achieving a SALT score of ≤20 was significantly greater in the baricitinib 2mg (33.3%) and 4mg (51.9%) groups versus placebo (3.6%) at week 3649. More recently, brepocitinib, a selective TYK‑2/JAK‑1 inhibitor, and ritlecitinib, a dual JAK3/TEC inhibitor, have demonstrated efficacy in treating AA in phase II clinical trials50.
While generally well tolerated, the most common adverse events associated with JAK inhibitor use are infection, nausea, transaminitis, lipid panel derangements and elevated CK11. Albeit rare, venous thromboembolic events have been reported. Similarly, an increased risk of malignancy associated with JAK inhibitors has been reported but appears to be comparable to that of DMARDs11.
Conclusions
AA is an unpredictable, relapsing and remitting disease that has a variable clinical course. Quality of life may be significantly compromised in sufferers. There is considerable variation in how AA is treated by dermatologists. As the therapeutic armamentarium continues to grow, so too does the need for standardised management guidelines. Severity assessment tools that adequately capture the psychosocial burden of disease will be critical in helping to identify candidates for current and emerging systemic therapies.
Author contribution
We declare that all listed authors have made substantial contributions to the conception of this work, drafting of this work and final approval of the version to be published. Furthermore, all authors attest to the validity and integrity of this work.
Acknowledgements
None.
Conflict of interest
The authors declare no conflicts of interest.
Ethics statement
An ethics statement is not applicable.
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Author(s)
Anthony Moussa*1, Laita Bokhari1 and Rodney D Sinclair1,2
1Sinclair Dermatology, East Melbourne, VIC, Australia
2University of Melbourne, Melbourne, VIC, Australia
Corresponding author Email anthonymoussa@outlook.com
References
- Gilhar A, Etzioni A, Paus R. Alopecia areata. N Engl J Med 2012 Apr 19;366(16):1515–25. doi:10.1056/NEJMra1103442. PMID:22512484.
- Mirzoyev SA, Schrum AG, Davis MDP, et al. Lifetime incidence risk of alopecia areata estimated at 2.1% by Rochester Epidemiology Project, 1990–2009. J Invest Dermatol 2014 Apr;134(4):1141–1142. doi:10.1038/jid.2013.464. Epub 2013 Nov 11. PMID:24202232; PMCID:PMC3961558.
- Pratt CH, King LE Jr, Messenger AG, et al. Alopecia areata. Nat Rev Dis Primers 2017 Mar 16;3:17011. doi:10.1038/nrdp.2017.11. PMID:28300084; PMCID:PMC5573125.
- Shi Q, Duvic M, Osei JS, et al. Health-related Quality of Life (HRQoL) in alopecia areata patients – a secondary analysis of the National Alopecia Areata Registry Data. J Investig Dermatol Symp Proc 2013;16(1):S49–50.
- Sinclair RD. Alopecia areata and suicide of children. Med J Aust 2014;200:145–145.
- Cranwell WC, Lai VW, Photiou L, et al. Treatment of alopecia areata: an Australian expert consensus statement. Australas J Dermatol 2019;60(2):163–70.
- Alkhalifah A, Alsantali A, Wang E, et al. Alopecia areata update: part I. Clinical picture, histopathology, and pathogenesis. J Am Acad Dermatol 2010 Feb;62(2):177–88, quiz 189–90. doi:10.1016/j.jaad.2009.10.032. PMID:20115945.
- Barahmani N, Schabath MB, Duvic M, et al. History of atopy or autoimmunity increases risk of alopecia areata. J Am Acad Dermatol 2009 Oct;61(4):581–91. doi:10.1016/j.jaad.2009.04.031. Epub 2009 Jul 16. PMID:19608295.
- Paus R, Bertolini M. The role of hair follicle immune privilege collapse in alopecia areata: status and perspectives. J Investig Dermatol Symp Proc 2013 Dec;16(1):S25–7. doi:10.1038/jidsymp.2013.7. PMID:24326544.
- Bertolini M, McElwee K, et al. Hair follicle immune privilege and its collapse in alopecia areata. Exp Dermatol 2020 Aug;29(8):703–725. doi:10.1111/exd.14155. PMID:32682334.
- Ismail FF, Sinclair R. JAK inhibition in the treatment of alopecia areata – a promising new dawn? Expert Rev Clin Pharmacol 2020 Jan;13(1):43–51. doi:10.1080/17512433.2020.1702878. Epub 2019 Dec 22. PMID:31865802.
- Villasante Fricke AC, Miteva M. Epidemiology and burden of alopecia areata: a systematic review. Clin Cosmet Investig Dermatol 2015 Jul 24;8:397–403. doi:10.2147/CCID.S53985. PMID:26244028; PMCID:PMC4521674.
- Tan E, Tay YK, Giam YC. A clinical study of childhood alopecia areata in Singapore. Pediatr Dermatol 2002 Jul–Aug;19(4):298–301. doi:10.1046/j.1525-1470.2002.00088.x. PMID:12220271.
- Lee H, Jung SJ, Patel AB, et al. Racial characteristics of alopecia areata in the United States. J Am Acad Dermatol 2020 Oct;83(4):1064–1070. doi:10.1016/j.jaad.2019.06.1300. Epub 2019 Jul 3. PMID:31279016.
- Finner AM. Alopecia areata: clinical presentation, diagnosis, and unusual cases. Dermatol Ther 2011 May–Jun;24(3):348–54. doi:10.1111/j.1529-8019.2011.01413.x. PMID:21689244.
- Atış G, Ferhatoğlu ZA. Trichoscopic clues for the diagnosis of alopecia areata. Turkderm-Turk Arch Dermatol Venereol 2020;54:76–8. doi:10.4274/turkderm.galenos.2020.00344
- Waśkiel A, Rakowska A, Sikora M, et al. Trichoscopy of alopecia areata: an update. J Dermatol 2018 Jun;45(6):692–700. doi:10.1111/1346-8138.14283. Epub 2018 Mar 22. PMID:29569271.
- Cervantes J, Fertig RM, Maddy A, et al. Alopecia areata of the beard: a review of the literature. Am J Clin Dermatol 2017 Dec;18(6):789–796. doi:10.1007/s40257-017-0297-6. PMID:28555441.
- Chelidze K, Lipner SR. Nail changes in alopecia areata: an update and review. Int J Dermatol 2018 Jul;57(7):776–783. doi:10.1111/ijd.13866. Epub 2018 Jan 10. PMID:29318582.
- Whiting DA. Histopathologic features of alopecia areata: a new look. Arch Dermatol 2003 Dec;139(12):1555–9. doi:10.1001/archderm.139.12.1555. PMID:14676070.
- Dy LC, Whiting DA. Histopathology of alopecia areata, acute and chronic: Why is it important to the clinician? Dermatol Ther 2011 May–Jun;24(3):369–74. doi:10.1111/j.1529-8019.2011.01414.x. PMID:21689247.
- Watanabe-Okada E, Amagai M, Ohyama M. Histopathological investigation of clinically non-affected perilesional scalp in alopecias detected unexpected spread of disease activities. J Dermatol 2014 Sep;41(9):802–7. doi:10.1111/1346-8138.12591. Epub 2014 Aug 25. PMID:25156442.
- Olsen EA, Roberts J, Sperling L, et al. Objective outcome measures: collecting meaningful data on alopecia areata. J Am Acad Dermatol 2018 Sep;79(3):470–478.e3. doi:10.1016/j.jaad.2017.10.048. Epub 2017 Nov 8. PMID:29128463; PMCID:PMC7450487.
- Olsen EA, Hordinsky MK, Price VH, et al. Alopecia areata investigational assessment guidelines – part II. National Alopecia Areata Foundation. J Am Acad Dermatol 2004 Sep;51(3):440–7. doi:10.1016/j.jaad.2003.09.032. PMID:15337988.
- Olsen EA, Canfield D. SALT II: a new take on the Severity of Alopecia Tool (SALT) for determining percentage scalp hair loss. J Am Acad Dermatol 2016 Dec;75(6):1268–1270. doi:10.1016/j.jaad.2016.08.042. PMID:27846957.
- Wyrwich KW, Kitchen H, Knight S, et al. The Alopecia Areata Investigator Global Assessment scale: a measure for evaluating clinically meaningful success in clinical trials. Br J Dermatol 2020 Oct;183(4):702–709. doi:10.1111/bjd.18883. Epub 2020 Apr 3. PMID:31970750; PMCID:PMC7586961.
- Jang YH, Moon SY, Lee WJ, et al. Alopecia Areata Progression Index, a scoring system for evaluating overall hair loss activity in alopecia areata patients with pigmented hair: a development and reliability assessment. Dermatol 2016;232(2):143–9. doi:10.1159/000442816. Epub 2016 Jan 13. PMID:26757319.
- Majid I, Sameem F, Sultan J, et al. Alopecia Areata Severity Index (AASI): a reliable scoring system to assess the severity of alopecia areata on face and scalp – a pilot study. J Cosmet Dermatol 2021 Aug;20(8):2565–2570. doi:10.1111/jocd.14289. Epub 2021 Jun 26. PMID:34129730.
- King BA, Mesinkovska NA, Craiglow B, et al. Development of the alopecia areata scale for clinical use: results of an academic–industry collaborative effort. J Am Acad Dermatol 2021 Aug 30:S0190-9622(21)02387-2. doi:10.1016/j.jaad.2021.08.043. Epub ahead of print. PMID:34474079.
- Wyrwich KW, Kitchen H, Knight S, et al. Development of Clinician-Reported Outcome (ClinRO) and Patient-Reported Outcome (PRO) measures for eyebrow, eyelash and nail assessment in alopecia areata. Am J Clin Dermatol 2020 Oct;21(5):725–732. doi:10.1007/s40257-020-00545-9. PMID:32803546; PMCID:PMC7473969.
- Stefanis AJ, Arenberger P, Arenbergerova M, et al. Alopecia barbae severity score: a novel scoring system to estimate the extent of beard loss and success of treatment. Br J Dermatol 2021 Oct;185(4):847–849. doi:10.1111/bjd.20489. Epub 2021 Jul 22. PMID:33997953.
- Petukhova L, Christiano AM. Functional interpretation of genome-wide association study evidence in alopecia areata. J Invest Dermatol 2016 Jan;136(1):314–317. doi:10.1038/JID.2015.402. PMID:26763452; PMCID:PMC4870380.
- Betz RC, Petukhova L, Ripke S, et al. Genome-wide meta-analysis in alopecia areata resolves HLA associations and reveals two new susceptibility loci. Nat Commun 2015 Jan 22;6:5966. doi:10.1038/ncomms6966. PMID:25608926; PMCID:PMC4451186.
- Fischer J, Degenhardt F, Hofmann A, et al. Genome wide analysis of copy number variants in alopecia areata in a Central European cohort reveals association with MCHR2. Exp Dermatol 2017 Jun;26(6):536–541. doi:10.1111/exd.13123. Epub 2017 Mar 23. PMID:27306922.
- Betz RC, Pforr J, Flaquer A, et al. Loss-of-function mutations in the filaggrin gene and alopecia areata: strong risk factor for a severe course of disease in patients comorbid for atopic disease. J Invest Dermatol 2007 Nov;127(11):2539–43. doi:10.1038/sj.jid.5700915. Epub 2007 Jun 21. PMID:17581619.
- Tanemura A, Oiso N, Nakano M, Itoi S, et al. Alopecia areata: infiltration of Th17 cells in the dermis, particularly around hair follicles. Dermatol 2013;226(4):333–6. doi:10.1159/000350933. Epub 2013 Jul 4. PMID:23838575.
- El-Morsy EH, Eid AA, Ghoneim H, et al. Serum level of interleukin-17A in patients with alopecia areata and its relationship to age. Int J Dermatol 2016 Aug;55(8):869–74. doi:10.1111/ijd.12994. Epub 2015 Oct 16. PMID:26475394.
- Loh SH, Moon HN, Lew BL, Sim WY. Role of T helper 17 cells and T regulatory cells in alopecia areata: comparison of lesion and serum cytokine between controls and patients. J Eur Acad Dermatol Venereol 2018 Jun;32(6):1028–1033. doi:10.1111/jdv.14775. Epub 2018 Jan 17. PMID:29283462.
- Ito T, Kageyama R, Nakazawa S, Honda T. Understanding the significance of cytokines and chemokines in the pathogenesis of alopecia areata. Exp Dermatol 2020 Aug;29(8):726–732. doi:10.1111/exd.14129. Epub 2020 Jul 3. PMID:32533873.
- Flanagan K, Sperling L, Lin J. Drug-induced alopecia after dupilumab therapy. JAAD Case Rep 2018 Dec 6;5(1):54–56. doi:10.1016/j.jdcr.2018.10.010. PMID:30560185; PMCID:PMC6289959.
- Messenger AG, McKillop J, Farrant P, et al. British Association of Dermatologists’ guidelines for the management of alopecia areata 2012. Br J Dermatol 2012 May;166(5):916–26. doi:10.1111/j.1365-2133.2012.10955.x. PMID:22524397.
- Fukuyama M, Ito T, Ohyama M. Alopecia areata: current understanding of the pathophysiology and update on therapeutic approaches, featuring the Japanese Dermatological Association guidelines. J Dermatol 2022 Jan;49(1):19–36. doi:10.1111/1346-8138.16207. Epub 2021 Oct 28. PMID:34709679.
- Singh G, Lavanya M. Topical immunotherapy in alopecia areata. Int J Trichol 2010 Jan;2(1):36–9. doi:10.4103/0974-7753.66911. PMID:21188022; PMCID:PMC3002409.
- Phan K, Ramachandran V, Sebaratnam DF. Methotrexate for alopecia areata: a systematic review and meta-analysis. J Am Acad Dermatol 2019 Jan;80(1):120–127.e2. doi:10.1016/j.jaad.2018.06.064. Epub 2018 Jul 10. PMID:30003990.
- Moussa A, Bokhari L, Sinclair RD. Systemic minoxidil as maintenance treatment in alopecia areata: a retrospective case series of 24 patients. Clin Exp Dermatol 2021 Dec 4. doi:10.1111/ced.15051. Epub ahead of print. PMID:34862632.
- Kerkemeyer KLS, Sinclair R. Treatment of chronic alopecia areata with tildrakizumab: an open-label pilot study. Int J Dermatol 2020 May;59(5):e136–e137. doi:10.1111/ijd.14826. Epub 2020 Mar 3. PMID:32124974.
- Speeckaert R, Lambert J, van Geel N. Learning from success and failure: biologics for non-approved skin diseases. Front Immunol 2019 Aug 8;10:1918. doi:10.3389/fimmu.2019.01918. PMID:31440261; PMCID:PMC6694799.
- Almutairi N, Nour TM, Hussain NH. Janus kinase inhibitors for the treatment of severe alopecia areata: an open-label comparative study. Dermatol 2019;235(2):130–136. doi:10.1159/000494613. Epub 2018 Dec 19. PMID:30566941.
- King B, Ko J, Forman S, et al. Efficacy and safety of the oral Janus kinase inhibitor baricitinib in the treatment of adults with alopecia areata: Phase 2 results from a randomized controlled study. J Am Acad Dermatol 2021 Oct;85(4):847–853. doi:10.1016/j.jaad.2021.05.050. Epub 2021 Jun 16. PMID:34090959.
- Peeva E, Guttman-Yassky E, Banerjee A, et al. Maintenance, withdrawal, and re-treatment with ritlecitinib and brepocitinib in patients with alopecia areata in a single-blind extension of a phase 2a randomized clinical trial. J Am Acad Dermatol 2021 Dec 13:S0190-9622(21)02964-9. doi:10.1016/j.jaad.2021.12.008. Epub ahead of print. PMID:34915057.

